|
Información || Congresos || Cursos || Territoriales || Noticias || Patología || Telepatología |
|
Información || Congresos || Cursos || Territoriales || Noticias || Patología || Telepatología |
| . |
[XXII Congreso Nacional (Palma de Mallorca)] [XXI Congreso Nacional (Madrid)] [ XX Congreso Nacional (Pamplona) ] [XIX Congreso Nacional (Barcelona) ] [XVIII Congreso Nacional (Málaga) ]
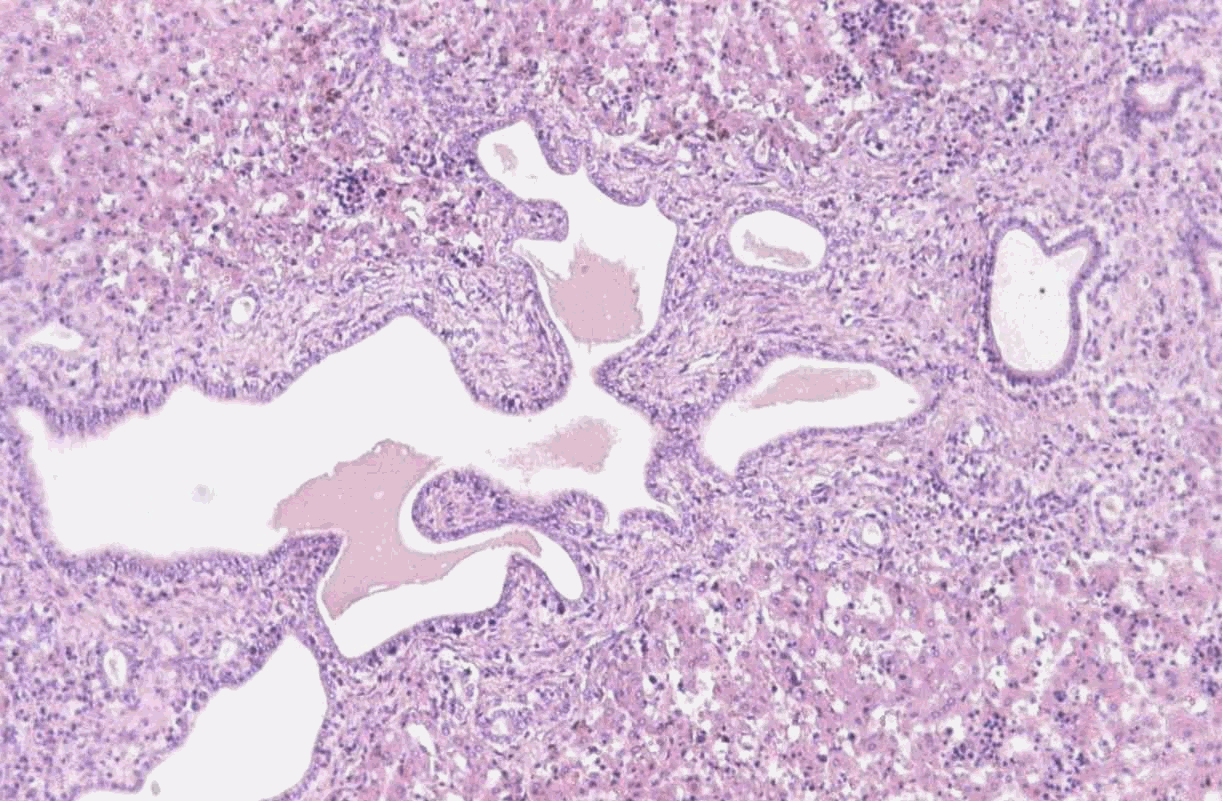
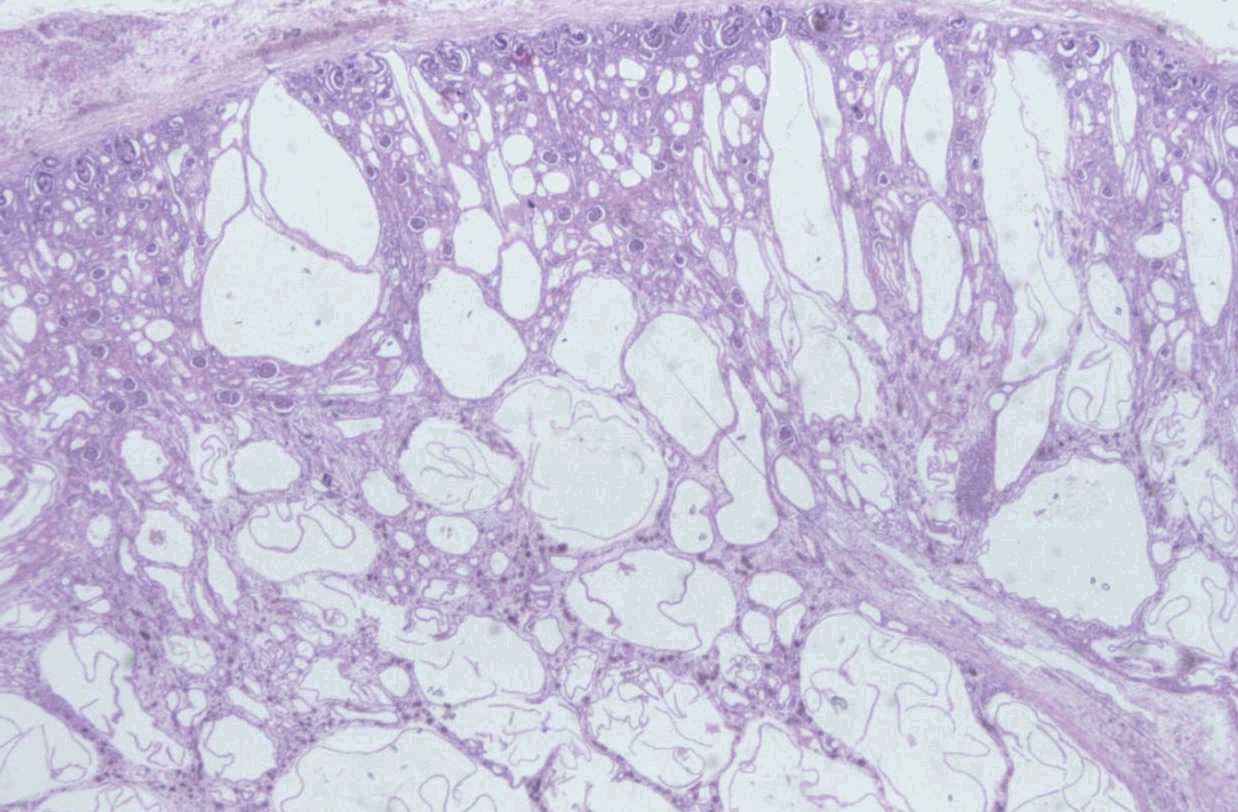
SEMINARIO DE PATOLOGÍA PEDIÁTRICA CASO Nº 2. Recién nacido prematuro de 33 semanas de gestación que, al nacer, presenta insuficiencia respiratoria grave que precisa reanimación enérgica y ventilación de alta frecuencia. La exploración muestra nefromegalia gigante. La radiografía de tórax muestra campos pulmonares muy pequeños, correspondientes a hipoplasia pulmonar. Fallece a los cinco días de vida por insuficiencia respiratoria. Se envía una muestra del tejido renal obtenido en la autopsia. DISCUSIÓN Las características macroscópicas y microscópicas de este caso corresponden a una Enfermedad Renal Quística Autosómica Recesiva (ARPKD), que ha recibido los nombres de poliquistosis hepato-renal de tipo infantil o poliquistosis renal tipo Potter I. Se caracteriza por afectación renal y hepática, con la presencia de grandes riñones poliquísticos, con dilatación longitudinal de túbulos renales, perpendiculares a la superficie renal y con conservación de la población de glomérulos (el número de nefronas es normal). La pelvis y los cálices renales son normales. En la ARPKD el primordio ureteral y el blastema metanéfrico muestran una diferenciación inicial normal pero se produce una anomalía en el desarrollo de los ductos colectores terminales que se extiende hasta la superficie cortical con dilataciones saculares o diverticulares de sus ramas inferiores. En el hígado se observa la imagen de una fibrosis hepática congénita (FHC) debida a una malformación de la placa ductal de los ductos biliares interlobulares que lleva a un trastorno de las interacciones inductivas epiteliomesenquimales y como consecuencia a la persistencia de un exceso de estructuras epiteliales ductales primitivas. Estos ductos biliares primitivos sufren una colangiopatía destructiva progresiva de evolución variable. Todos los casos de ARPKD presentan FHC, pero solamente el 50% de FHC se asocian a ARPKD. El locus del gen responsable de ARPKD se halla en 6p21.1-p12. Debido a la variabilidad en la presentación clínica de esta enfermedad se distinguieron cuatro grupos clínico-patológicos: perinatal, neonatal, infantil y juvenil, según la edad de presentación y la gravedad de los síntomas hepáticos y renales, que sugerían cuatro entidades genéticas diferentes. Posteriormente la evidencia de variabilidad fenotípica intrafamiliar y los estudios de ligamiento genético sugieren que esta variabilidad fenotípica es debida a una heterogeneidad alélica, comprobada por el análisis mutacional descrito por Ward et al (2002). En los casos de presentación perinatal no suelen plantearse problemas diagnósticos en el examen de autopsia. Pero sí pueden plantearse en caso de autopsia de fetos de segundo trimestre, principalmente al principio del segundo trimestre, en que las alteraciones se encuentran en un período inicial y pueden ser menos evidentes. El diagnóstico diferencial de la patología renal quística debe incluir: -Displasia renal: se plantea el diagnóstico diferencial en aquellos casos de nefromegalia bilateral con displasia renal bilateral, de quistes pequeños. La displasia renal es un trastorno de la diferenciación metanéfrica. En estos casos el tejido renal se halla desorganizado, la pelvis renal es atrésica, los quistes son irregulares y existe importante afectación de los glomérulos. Se han descrito casos de displasia renal bilateral asociada a fibrosis hepática congénita con afectación pancreática, de herencia autosómica recesiva. Alteraciones similares pueden también encontrarse en la trisomía 9, síndrome de Meckel, condrodisplasia de Jeune, de Saldino-Noonan y aciduria glutárica tipo II. Una vez descartados los síndromes identificables, los casos restantes de displasia renal-hepática-pancreática no constituyen necesariamente un grupo homogéneo. -Enfermedad renal poliquística dominante (ADPKD, Riñón poliquístico tipo adulto o Potter II): En estadios presintomáticos en pacientes jóvenes o fetos, se observan quistes redondeados de tamaño variable derivados de cualquier zona de la nefrona o de los ductos colectores, de distribución irregular, con prominenete ectasia del espacio de Bowmannun tipo de enfermedad glomeruloquística). Se han descrito 2 genes responsables: PKD1 (16p13.3) y PKD2 (4p13-q23). Aunque cursa con quistes hepáticos de distribución focal, se han descrito algunos casos en que se hallaba asociada a fibrosis hepática congénita. En estas familias la ARPKD se transmite de forma dominante pero la FHC no. -Enfermedad gloméruloquística: es un grupo heterogéneo de enfermedades que cursan con quistes glomerulares en que se encuentran la ADPKD de inicio precoz , el sindrome cerebro-hepato-renal de Zellweger, esclerosis tuberosa, trisomía 13, síndrome de costilla corta-polidactilia tipo Majewski, síndrome oro-facial-digital, síndrome braquimesomelia-renal y algunos casos de displasia renal. En la infancia el 50% aproximadamente corresponden a ADPKD de inicio precoz. -Quistes renales asociados a síndromes: además de los ya descritos en el apartado anterior hay que tener en cuenta los síndromes de translocación cromosómica, síndromes de costilla corta-polidactilia, Ehler-Danlos, síndrome de lisencefalia, distrofia torácica asfixiante de Jeune, síndrome de Meckel y sus variantes. La afectación renal varía desde la presencia de quistes aislados a una displasia quística difusa y, en algunos síndromes, se acompaña de disgenesia de ductos biliares similar a la que se observa en los casos de ARPKD. -En la enfermedad quística medular se distinguen el riñón medular en esponja y la nefronoptisis familiar-enfermedad medular quística que, cuando se presenta en la infancia, puede ser de herencia autosómica recesiva.
BIBLIOGRAFÍA: -Bernstein J, Risdom R A, Gilbert-Barness E. Renal system in Potters Pathology of the Fetus and Infant. Ed by Enid Gilbert-Barness. Mosby-Year Book Inc 1997; Chapter 22 p 880-888. -Kaczorowski JM, Halterman J, Spitalnik P, Mannick E, Chin-To Fong. Congenital hepatic fibrosis and autosomal dominant polycystic kidney disease. Pediatric Pathology and Molecular Medicine 2001; 20:245-248. -Ward C J, Hogan M C, Rossetti S, Walker D, Sneddon T, Wang X, Kubly V, Cunningham J M, Bacallao R, Ishibashi M, Milliner D S, Torres V E, Harris P C. The gene mutated in autosomal recessive polycystic kidneu dosease encodes a large, receptor-like protein. Nature Genet 2002; 30: 259-269.
[Programa del Congreso] [Índice del Seminario] [Cursos Cortos] [Seminarios]
|
. | |||
| © SEAP. Sociedad Española de Anatomía Patológica | Actualizado: 09/07/2003 |
||||