Poster
# 110
![]()
Poster |
|
6th Internet World Congress for Biomedical Sciences |
Trevor Sharp(1), Elena Castro(2)
(1)Department of Clinical Pharmacology. University of Oxford - Oxford. United Kingdom
(2) Clinical Pharmacology. University of Oxford - Oxford. United Kingdom
|
|
|
|
|
|
[Pharmacology] |
[Psychiatry] |
It is well established that selective serotonin reuptake inhibitors (SSRI) are effective in the treatment of major depression, however, as with other antidepressants, several weeks of treatment are necessary before their full therapeutic effect becomes apparent. SSRIs may not be clinically effective at the start of treatment because the drugs cause indirect activation of presynaptic (somatodendritic) 5-HT1A autoreceptors within the midbrain raphe nuclei. This reduces 5-HT neuronal firing and decreases 5-HT release in the terminal regions (1,8).
Animal data show that selective antagonists of the 5-HT1A receptor potentiate the effects of the SSRIs on presynaptic 5-HT function by preventing 5-HT1A autoreceptor activation (7). On this basis clinical trials have tested the utility of the b-adrenoceptor/5-HT1A ligand pindolol (specifically, (±)-pindolol) in SSRI augmentation. Significantly, pindolol/SSRI combinations have been found to produce a more rapid onset of antidepressant action in some although not all studies (11).
A factor complicating the use of 5-HT1A antagonists in depression is that in addition to blocking the presynaptic 5-HT1A receptor, these drugs will also block the postsynaptic 5-HT1A receptor, an effect which could interfere with the antidepressant effect. In the case of pindolol (specifically, (-)-pindolol), however, there is animal data indicating that it is presynaptic selective (15) but this is not confirmed in other studies (6). Pindolol is known to have high affinity for 5-HT1A sites in membrane preparations of rat and human brain as well as cells transfected with the human 5-HT1A receptor (9,12) but information on the affinity of pindolol at presynaptic and postsynaptic 5-HT1A receptors is needed.
Here we have determined the affinity of (±)-pindolol for different populations of native 5-HT1A receptors in postmortem human and rat brain using receptor autoradiography and the selective 5-HT1A radioligand [3H]WAY-100635.
Animal brains were obtained from adult male rats (250-275 g, Sprague-Dawley, Harlan-Olac, Bicester, U.K.) and stored at -20°C. Human brain tissue was obtained at autopsy from four subjects (two men and two women, mean age=67±9 years, postmortem delay=24.8±3.6 h) with no history of neurological or psychiatric disorders. Sequential 14 µm cryostat sections from hippocampus and dorsal raphé nucleus (DRN) of each rat and human brain were thaw-mounted on to gelatinised slides and stored at -20°C until use.
5-HT1A receptor binding was determined autoradiographically using [3H]WAY-100635. Brain sections were preincubated for 30 min in 50 mM Tris-HCl buffer (pH 7.5). For the inhibition studies, sections were then incubated for 2 h in either 50 mM Tris-HCl buffer containing 10 µM pargyline with 3 nM [3H]WAY-100635 in the presence or absence of (±)-pindolol at 7 different concentrations (0.1 nM-1 µM). Each drug concentration was tested in triplicate (rat) or duplicate (human). Inhibition curves for the DRN were always matched with inhibition curves for the corresponding hippocampus.
Non-specific binding was determined using 10 µM unlabeled 5-HT. In saturation studies, conditions used were identical to those described above, except that the sections were incubated with increasing concentrations of [3H]WAY-100635 (0.25 - 12 nM).
Following incubation, sections were washed for 2 X 2 min in ice-cold buffer, briefly dipped in de-ionised water at 4°C, and then air-dried. Autoradiograms were generated by apposing labelled tissues to microscale tritium standards and tritium-sensitive film (Hyperfilm, Amersham plc, UK) for 6 weeks. Autoradiograms were quantified using a computerized image analysis system (MCID system, Image Research, St. Catharine´s, Canada).
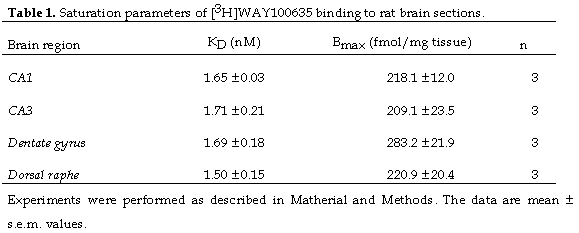
Ki values were calculated using the IC50 values determined in the inhibition studies and KD values calculated in saturation studies. For rat tissue, we used the KD of [3H]WAY-100635 estimated in our saturation experiments (1.5-1.7 nM). For human tissue, we used the KD previously determined for human hippocampus by Burnet et al. (3). The data were paired in that each Ki value for the DRN was matched with the Ki value of the corresponding hippocampus.
Data are presented as mean ± s.e.m. values. Binding data were analysed using the programme InPlot (GraphPad Software). Ki values were compared across species and brain areas using one-way ANOVA followed post hoc by Dunnett´s t-test.
[3H]WAY-100635 (specific activity 80 Ci/mmol) was supplied by Wyeth Research UK Ltd. The following drugs were used: (±)-pindolol HCl (Sigma), 5-HT creatinine sulphate (Sigma) and pargyline (Sigma).
Scatchard and nonlinear regression analysis revealed that [3H]WAY-100635 interacted with a single class of binding sites in rat brain. This binding was saturable, specific and of high affinity in DRN (KD= 1.5 nM) and all the areas of the hippocampus tested, specifically CA1, CA3 and dentate gyrus (KD= 1.65-1.71 nM). The total number of binding sites in the DRN were of the same order of those in the hippocampal areas Table 1. At the concentration of [3H]WAY-100635 used in inhibition assays (3 nM) non-specific binding, as defined by 10 µM 5-HT, represented less than 10% of the total binding (data not shown).
(±)-Pindolol caused a concentration-dependent inhibition of [3H]WAY-100635 in all rat brain areas tested and the competition curves were best described by a single binding site with Hill coefficients close to unity.
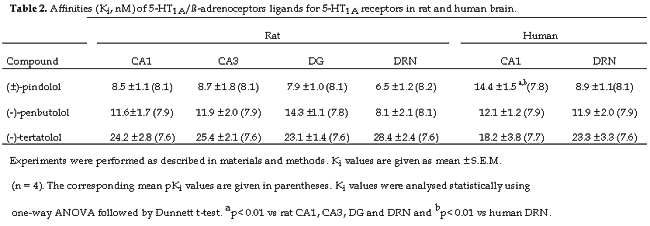
Autoradiograms illustrating the inhibition by (±)-pindolol of [3H]WAY-100635 binding in the rat DRN and hippocampus are shown in figure 1. (±)-Pindolol did not discriminate between pre- and postsynaptic 5-HT1A binding sites. Thus, the inhibition curves for (±)-pindolol obtained from CA1, CA3 and dentate gyrus of the rat hippocampus produced Ki values (7.9- 8.7 nM) were not statistically different from the Ki value of the DRN (6.5 nM) (Table 2).
Human brain sections incubated with [3H]WAY-100635 revealed substantial 5-HT1A receptor binding in the DRN and CA1 of the hippocampus, with considerably weaker binding in CA3 and dentate gyrus. Due to the relatively weak signal observed in these later two regions, inhibition experiments were quantified only within CA1.
As with the rat, (±)-pindolol, demonstrated high affinity for the 5-HT1A binding sites in human hippocampus and DRN figure 2, and the competition curves for (±)-pindolol were best described by a single binding site. In contrast to the rat, (±)-pindolol showed a small degree of selectivity for the pre- versus postsynaptic 5-HT1A binding sites in the human. Thus, the Ki value of (±)-pindolol for the human DRN (8.9 nM) was lower than that for the CA1 of hippocampus (14.4 nM) and this difference was statistically significant (Table 2).
Drugs which block presynaptic (somatodendritic) 5-HT1A receptors are candidates for testing as adjuncts to SSRIs to improve the treatment of severe depressive illness. The purpose of this receptor autoradiographic study was to determine the affinity of the b-adrenoceptor/5-HT1A ligand (±)-pindolol for pre- and postsynaptic 5-HT1A receptors in rat and human brain. Using the selective antagonist radioligand [3H]WAY-100635, our results show that (±)-pindolol has high affinity for presynaptic (DRN) and postsynaptic (CA3, CA1 and dentate gyrus in rat and CA1 in human) 5-HT1A receptors in both species. In the rat (±)-pindolol did not show preference for the presynaptic receptor although in the human the drug showed a significant though modestly higher affinity for the 5-HT1A site in the DRN versus CA1.
A cross species (rat, human, guinea pig, primate) study of the affinity of (-)-pindolol at pre- and postsynaptic 5-HT1A receptor was recently reported by Raurich et al., (14). The latter study found that (-)-pindolol did not discriminate between 5-HT1A sites in the DRN and hippocampus which is in broad keeping with the present data. Our finding of a slightly higher affinity of (±)-pindolol for the human DRN could therefore be a chance finding. However, the fact that we made within subject measurements of drug affinity at pre- and postsynaptic sites adds to the power of our experimental design and would increase the probability of detecting small changes. Clearly, the issue will only be resolved definitively by the study of a larger number of postmortem human brains.
The finding that (±)-pindolol has equal affinity at the rat pre- and postsynaptic 5-HT1A receptor agrees with the results of a recent electrophysiological study which found that (-)-pindolol blocked 5-HT1A agonist-induced electrophysiological responses in the rat DRN and hippocampus with equal potency (6). These question the original idea that pindolol is ineffective at blocking postsynaptic 5-HT1A receptors in hippocampus (15). The latter conclusion was derived from experiments in which systemically administered pindolol did not block the inhibition of hippocampal neurones by microiontophoretically applied 5-HT1A agonist. In the light of the newer findings, it may be the 5-HT1A antagonist property of pindolol was not adequately assessed in the study of Romero et al., possibly because agonists were applied locally in too high concentrations.
Interestingly, despite the evidence that the affinity of pindolol for pre- and postsynaptic 5-HT1A receptors is similar if not the same, recent PET imaging studies using the ligand [11C]WAY-100635 report that (±)-pindolol displays a slight but consistent preferential occupancy for the pre- versus postsynaptic 5-HT1A receptor in both human and rat brain in vivo (10,13). Rather than being explained by affinity differences, these PET data might be explained by evidence that in vivo (±)-pindolol causes a fall in endogenous 5-HT in the terminal areas (5). Thus, as a consequence of decreased extracellular 5-HT a greater number of PET ligand molecules would occupy the postsynaptic 5-HT1A receptors and a higher dose of pindolol would be needed to displace them (10). Whatever the explanation, the PET studies indicate that the preferential occupancy of presynaptic 5-HT1A receptors by (±)-pindolol covers a narrow dose range that may be difficult to achieve in routine clinical practice (13).
The augmentation of SSRIs by drugs blocking the presynaptic 5-HT1A receptor could be compromised by simultaneous blockade of the postsynaptic 5-HT1A receptor, if it is assumed that activation of the latter receptor mediates the antidepressant effect. However, the latter assumption is based largely on indirect evidence: e.g. that antidepressant drugs and ECS cause an adaptive increase in postsynaptic 5-HT1A receptor function (2). Recent immediate early gene experiments from this laboratory certainly indicate that SSRI/5-HT1A antagonists in combination increase postsynaptic 5-HT receptor function (4), but through 5-HT receptors that are not of the 5-HT1A subtype. Increased transmission through non-5-HT1A receptors could therefore contribute to an antidepressant effect.
In conclusion, our results show that the putative SSRI augmenting agent, (±)-pindolol, has high affinity for both presynaptic and postsynaptic 5-HT1A receptors in postmortem human and rat brain. Our data do not support the idea that pindolol has marked selectivity for any of the 5-HT1A receptor populations studied in vitro. However, recent PET imaging studies in human volunteers indicate that over a narrow dose range, pindolol may preferentially occupy presynaptic 5-HT1A receptors in vivo (13). Whether this property of pindolol confers therapeutic advantages (ie. in major depression) over other 5-HT1A receptor antagonists currently available for clinical use (eg. tertatolol and penbutolol), awaits further investigation.
This work was supported by a Programme grant from the Medical Research Council (TS) and a travelling fellowship from the Spanish Government (MEC).
|
|
|
|
|
|
[Pharmacology] |
[Psychiatry] |
{kind=link}
{kind=link}