Poster
# 93
![]()
Poster |
|
6th Internet World Congress for Biomedical Sciences |
Yasuji Matsuoka(1), Mitsuhiro Okazaki(2), Yuko Sekino(3), Yoshihisa Kitamura(4)
(1)Nathan Kline Inst. - Orangeburg. United States
(2)(4)Dept Neurobiol. Kyoto Pharm Univ - Yamashina. Japan
(3)Department of Neurobiology and behavior. Gunma University School of Medicine - Maebashi. Japan
|
|
|
|
|
|
[Cell Biology & Cytology] |
[Neuroscience] |
Adenosine is a purine nucleotide with diverse biological functions. Purinergic receptors can be divided into two subgroups based on their agonist preference for adenosine (P1) or ATP (P2) (Fredholm et al., 1994). Adenosine receptors, also referred to as P1 purinoceptors, are members of the G protein-coupled receptor family that mediate the physiological effects of adenosine. The A1, A2a, A2b, and A3 adenosine receptor subtypes have been cloned (Olah and Stiles, 1995). These adenosine receptor subtypes show distinct distributions, and the A1 and A2a subtypes are expressed predominantly in the CNS (Fredholm et al., 1994). In the CNS, adenosine modulates synaptic transmission by blocking neurotransmitter release (Hollins and Stone, 1980; Ribeiro and Sebastiao, 1984), modulates long-term potentiation in the hippocampus (de Mendonca and Ribeiro, 1994), and affects cerebral blood flow (Phillis and Wu, 1981). Adenosine A1 receptors are known to mediate the suppression of neuronal activity in the hippocampus (Dunwiddie and Fredholm, 1989).
Cerebral ischemia results in the release of large amounts of excitatory amino acid transmitter, such as glutamate, into the extracellular space. This release is mirrored by a drastic increase in the extracellular concentration of adenosine (Globus et al., 1988; Hagberg et al., 1987). In addition, adenosine analog blocks experimentally-induced seizures in animals (Barraco et al., 1984), and electrical stimulation along with adenosine receptor antagonist induces status epilepticus (Young and Dragunow, 1995). Therefore, adenosine may act as an endogenous anticonvulsant (Dragunow et al., 1985; During and Spencer, 1992). Furthermore, binding to the A1 adenosine receptor is decreased in human temporal lobe epilepsy (Glass et al., 1996). Although these results strongly suggest that adenosine may participate in seizure and may exert neuroprotection, the role of endogenous adenosine in cell vulnerability has not yet been examined.
Kainic acid (KA) is a potent agonist at excitatory amino acid receptor subtypes in the CNS. Intracerebroventricular (i.c.v.) injection of KA induces selective neuronal loss in the CA3 subfield and activates glial cells in the rat hippocampus (Nalder et al., 1980). In addition, selective neuronal vulnerability has been reported; i.e., hypoxia, a main cause of ischemia, induced neurodegeneration in the CA3, but not in the CA1 (Taniguchi et al., 1994; Matsuoka et al., 1995, 1997a and 1997b), although the CA1 is vulnerable to ischemic stimuli (Kirino, 1982). Evidence for the neurodegenerative mechanisms of glutamate has accumulated (Dykens et al., 1987; Coyle and Puttfarcken, 1993). However, the molecular and cellular events responsible for the selective vulnerability of this population of neuronal cells to KA-induced seizure activity are not yet understood. Therefore, we tried to clarify the mechanism of selective neuronal vulnerability in the hippocampus by focusing on the A1 adenosine receptor in an animal model of KA-induced neurodegeneration, using microtubule associated protein-2 (MAP-2)-; phosphorylated c-Jun-; and CD11b-, glial fibrillary acidic protein (GFAP)-, and major histocompatibility complex (MHC) class II-immunoreactivities as markers for neuronal cell loss, neuronal apoptosis, and glial activation, respectively.
Animals:
Male Wistar rats (Crj: Wistar, Charles River, Atsugi, Japan) weighing approximately 300 g were used.
Materials:
-8-Cyclopenthyltheophylline (CPT) and N6-cyclopenthyladenosine (CHA) (A1 adenosine receptor antagonist and agonist, respectively): RBI
-KA: Sigma.
-Mouse monoclonal antibodies against rat MAP-2 and porcine GFAP were from Chemicon International, and rat CD11b and rat MHC class II were from Harlan Sera-Lab.
-Rabbit polyclonal antibodies against c-Jun and Ser73-phosphorylated c-Jun were from New England BioLabs. This anti-c-Jun antibody was raised using a synthetic peptide corresponding to amino acids 1-79 of human c-Jun. The anti-phosphorylated c-Jun antibody was raised using a synthetic phospho-Ser73 peptide corresponding to amino acids 68-77 (LLKLAS*ELERC) of human c-Jun (asterisk indicates phosphorylation). This anti-phosphorylated c-Jun antibody detects phosphorylated c-Jun at Ser73, yet will not react with nonphosphorylated c-Jun.
Drug administration, animal model, and experimental protocol
-CPT was injected i.p. 60 min before KA injection.
-CHA was co-injected with KA.
-One Êg KA was injected into the right lateral ventricle.
-As a control, rats were injected with the same amount of vehicle.
-Each group consisted of five rats.
Immunohistochemistry
-Twelve hours or 4 days after treatment, the animals were perfused with phosphate-buffered saline, followed by a cold fixative consisting of 4% paraformaldehyde in phosphate buffer.
-After perfusion, the brain was quickly removed and post-fixed for 24 hours.
-The free floating sections were incubated with antibodies against MAP-2 (1:3,000), c-Jun (1:200), phospho-c-Jun (1:200), CD11b (1:6,000), GFAP (1:5,000) or MHC class II (1:2,000) for 4 days at 4 C.
-The antibody was detected by an ABC Elite kit.
Quantitative analysis and statistical evaluation
-Immunostained sections (2.56 mm caudal from the bregma) were scanned using a camera (KY-F55MD, Victor, Tokyo, Japan) and then analyzed (WinRoof, Mitani Corp., Fukui, Japan).
-For the quantitative analysis of neurodegeneration, the percent of the MAP-2-immunoreactive area of each subfield was measured and used as an index of neuronal survival (decrement indicates neuronal cell loss). The percent of MAP-2-immunoreactive area was calculated by following formula: (MAP-2-immunoreactive area [%]) = (MAP-2-immunoreactive area [mm2]) / (area of subfield, i.e., CA1 or CA3 [mm2]).
-The number of c-Jun- and phosphorylated-c-Jun-immunoporeactive cells was counted in each subfield, i.e., CA1 or CA3, and presented as the number of immunopositive cells in each milisquare [cells/mm2].
-Data are presented as the mean } standard error of five rats. The significance of differences was determined by an analysis of variance (ANOVA) with the Bonferroni/Dunn posthoc test (Stat View, Abacus Concepts, Berkeley, CA).
Effect of A1 adenosine receptor antagonist for:
--Neuronal Cells.
--Glial Cells.
--Neuronal Apoptosis.
Effect of A1 adenosine receptor ANTAGONIST against KA-induced neurodegeneration
MAP-2-immunoreactivity was widely observed in vehicle-injected rat hippocampus (Fig. 1A). CPT itself appeared not to have any effect on MAP-2-immunoreactivity (Fig. 1B). After the i.c.v. injection of KA, MAP-2-immunoreactivity was lost specifically in the CA3 (Fig. 1C). Pretreatment with CPT (10 mg/kg, i.p.), an A1 adenosine receptor antagonist, at 60 min before the injection of KA, resulted in a marked reduction in MAP-2-immunoreactivity in both CA1 and CA3 (Fig. 1D). Quantitatively, the MAP-2-immunoreactive area was significantly decreased only in the CA3 after the injection of KA alone (Fig. 2B). It did not change in the CA1 (Fig. 2A). In contrast, the combination of KA and CPT (KA/CPT), induced a significant loss of MAP-2-immunoreactive area in the CA1 (Fig. 2).
Effect of A1 adenosine receptor agonist AGONIST KA-induced neuronal cell loss
The coadministration of CHA, an A1 adenosine receptor agonist, with KA attenuated the loss of MAP-2-immunoreactivity in the CA3 induced by KA (Fig. 1E). In addition, the coadministration of CHA with KA/CPT also attenuated the neuronal cell loss in both the CA1 and the CA3 (Fig. 1F). Quantitatively, the loss of the MAP-2-immunoreactive area in the CA3 with the injection of KA alone was significantly attenuated by the administration of CHA (Fig. 3B). The administration of CHA significantly attenuated the loss of the MAP-2-immunoreactive area in both the CA1(Fig. 3A) and the CA3 (Fig. 3B) caused by the injection of KA/CPT.
**Jump to the Top of Page, Fig. 1, Fig. 2, Fig. 3, Fig. 4, Fig. 5, Fig. 6, Fig. 7
Fig. 1: Effects of an A1 adenosine receptor antagonist (CPT) and agonist (CHA) on KA-induced neurodegeneration.
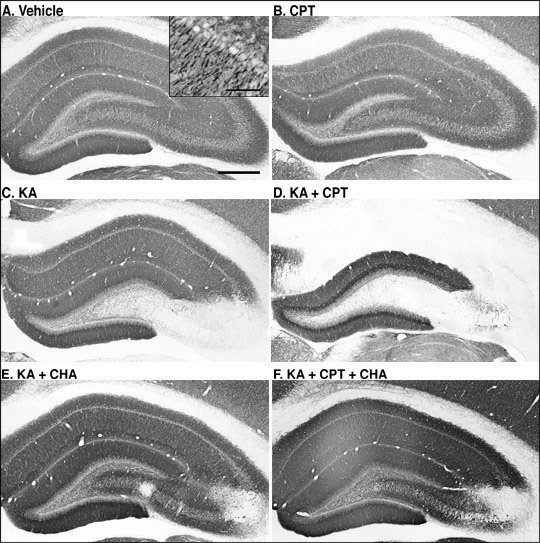
MAP-2-immunostained sections in the hippocampus 4 days after the injection.
**Jump to the Top of Page, Fig. 1, Fig. 2, Fig. 3, Fig. 4, Fig. 5, Fig. 6, Fig. 7
Fig. 2: Quantitative analysis of KA-induced neuronal cell loss and the effect of an A1 adenosine receptor antagonist (CPT)
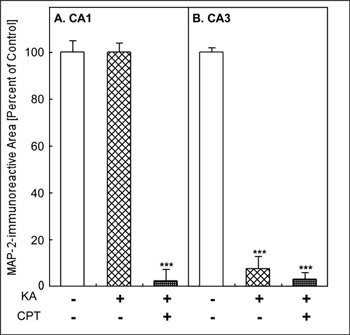
**Jump to the Top of Page, Fig. 1, Fig. 2, Fig. 3, Fig. 4, Fig. 5, Fig. 6, Fig. 7
Fig. 3: Quantitative analysis of KA- or KA/CPT-induced neuronal cell loss and the effect of an A1 adenosine receptor agonist (CHA).
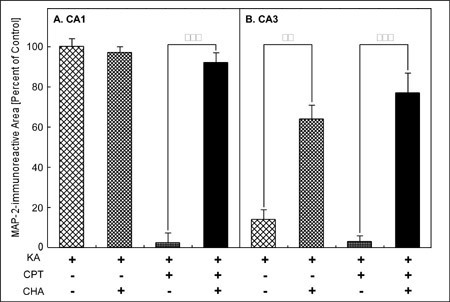
**Jump to the Top of Page, Fig. 1, Fig. 2, Fig. 3, Fig. 4, Fig. 5, Fig. 6, Fig. 7
Histological changes of after coadministration of KA, CPT, and/or CHA
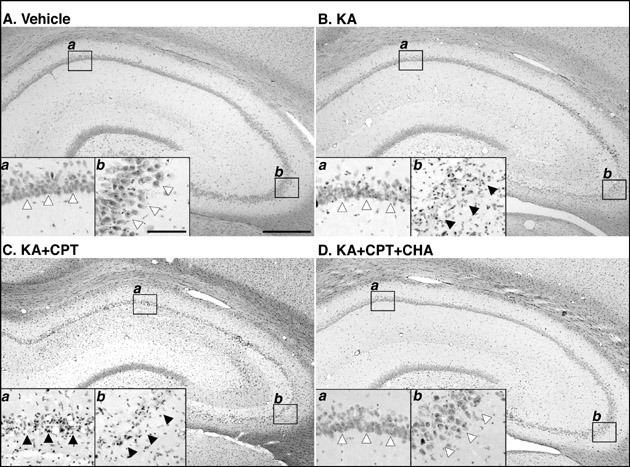
Injection of KA alone induced loss of the pyramidal neurons in CA3 (Fig. 4Bb), but not CA1 (Fig. 4Ba), although vehicle appeared not to have any effect on neurons (Fig. 4A). Pretreatment of CPT at 60 min before the injection of KA induced marked loss of neurons in both CA1 and CA3 (Fig. 4C). The coadministration of CHA with KA/CPT attenuated the neuronal cell loss in both the CA1 and the CA3 (Fig. 4D). Therefore, the histological changes, which examined using H-E staining, were conformity well with changes of MAP-2-immunoreactivity.
Fig. 4: Histological changes (hematoxylin and eosin staining)of rat hippocampal formation 4 days after injection of KA and effects of an A1 adenosine receptor antagonist (CPT) and agonist (CHA)
**Jump to the Top of Page, Fig. 1, Fig. 2, Fig. 3, Fig. 4, Fig. 5, Fig. 6, Fig. 7
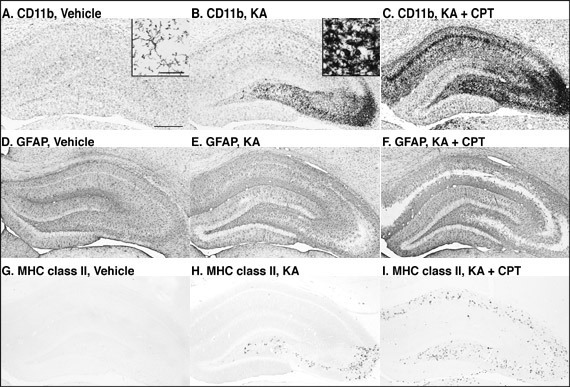
Changes in glial cells after KA-injection and the effect of adenosine receptor antagonist
Microglia (CD11b-immunostaining) have thin and longer processes that are typically ramified (Fig. 4A). Four days after the i.c.v. injection of KA, numerous microglia with thick and shorter processes, i.e., typical ameboid type, were observed in the CA3 (Fig. 4B inset), while the microglia in the other hippocampal subfields were mostly ramified (Fig. 4B). After treatment with KA/CPT, ameboid microglia with thick and shorter processes were also observed in the CA1 (Fig. 4C). Astrocytes (GFAP-immunostaining) were slightly activated by injection with KA (Figs. 4D-4F). MHC class II-immunoreactivity was undetectable in the vehicle-injected rat hippocampus (Fig. 4G). Four days after KA-injection, MHC class II-immunoreactivity was observed only in the CA3 (Fig. 4H). Treatment with KA/CPT also induced MHC class II-immunoreactivity in the CA1 (Fig. 4I). The regions in which glial activation occurred correlated well with those that showed neurodegeneration, as judged by MAP-2-immunoreactivity.
Fig. 5: Photomicrographs of CD11b-, GFAP-, and MHC class II-immunostained sections in the hippocampus 4 days after the injection.
**Jump to the Top of Page, Fig. 1, Fig. 2, Fig. 3, Fig. 4, Fig. 5, Fig. 6, Fig. 7
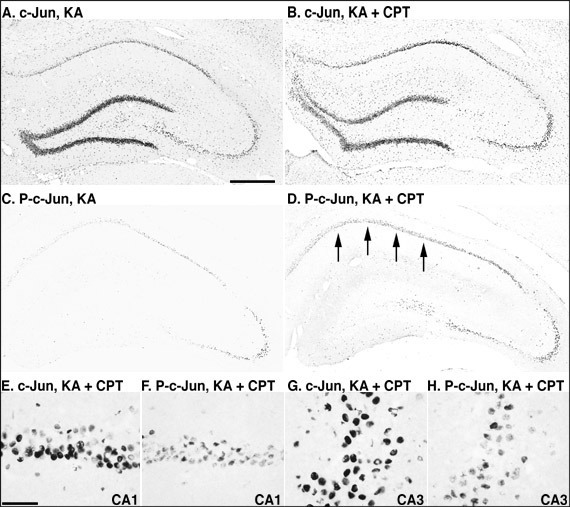
Phosphorylation of c-Jun after KA-injection and the effects of an adenosine receptor antagonist and agonist
In the vehicle-injected rat hippocampus, c-Jun was detected in the dentate gyrus, while phosphorylated c-Jun was not detected in the rat hippocampus. Twelve hours after the injection of KA or KA/CPT, c-Jun was markedly induced throughout the hippocampus (Figs. 5A and 5B). In contrast, c-Jun phosphorylation was detected dominantly in CA3 pyramidal neurons, and slightly in CA1 (Fig. 5C). When CPT was injected 60 min before KA injection, c-Jun phosphorylation was markedly induced in the CA1 (Fig. 5D). c-Jun phosphorylation was observed in some c-Jun-immunoreactive pyramidal neurons in both the CA1 (Figs. 5E and 5F) and the CA3 (Figs. 5G and 5H).
Quantitatively, treatment with KA alone or KA/CPT significantly increased the number of c-Jun immunoreactive cells in the CA1 and the CA3 (Fig. 6A). Although phosphorylated c-Jun was not detected in the CA1 or the CA3, phosphorylated c-Jun significantly increased after the injection of KA alone or KA/CPT in both the CA1 and CA3 (Fig. 6B). The phosphorylation of c-Jun in the CA3 was greater than that in the CA1 after the injection of KA alone. c-Jun phosphorylation was significantly enhanced in the CA1 by the injection of KA/CPT in comparison to that with KA alone.
Fig. 6: Photomicrographs of c-Jun and phosphorylated-c-Jun- (P-c-Jun) immunostained sections in the hippocampus 12 hours after the injection of KA or the combination of KA and CPT.
**Jump to the Top of Page, Fig. 1, Fig. 2, Fig. 3, Fig. 4, Fig. 5, Fig. 6, Fig. 7
Fig. 7: Quantitative analysis of the number of c-Jun- and phosphorylated-c-Jun-immunoreactive cells 12 hours after the injection of KA with or without CPT.
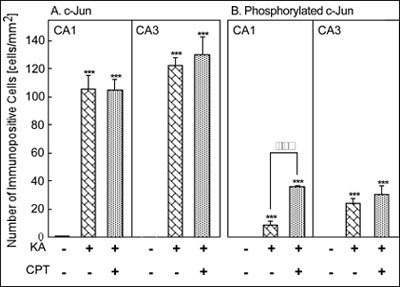
***P<0.001 versus vehicle injection, and õõP<0.01 versus injection of KA alone using ANOVA.
**Jump to the Top of Page, Fig. 1, Fig. 2, Fig. 3, Fig. 4, Fig. 5, Fig. 6, Fig. 7
Although the CA1 was not damaged by the injection of KA alone, the addition of an A1 adenosine receptor antagonist, CPT, exacerbated neuronal damage in the hippocampus, i.e., neuronal cell loss also occurred in the CA1. Microglia have thin and longer processes that were typically ramified in both non-treated and vehicle-injected rat brain. However, microglia were activated and showed a typical ameboid type morphology after the injection of KA or KA/CPT. In addition, MHC class II antigen was detected in the region of neuronal cell loss, but was not detected in the vehicle-injected rat brain. Several recent papers have reported that KA neurotoxicity and ischemia induce MHC class II-immunoreactive ameboid microglia (Akiyama et al., 1988; Finsen et al., 1993; Matsuoka et al., 1998b). In addition, reactive microglia that express MHC class II have been observed phagocytosing degenerated neuronal elements in Alzheimer´s disease, Parkinson´s disease, acquired immunodeficiency syndrome, and other neuronal degenerative disorders of humans (Dickson et al., 1993; McGeer et al., 1993). Therefore, the induction of MHC class II antigen suggests the strong activation of glial cells. The regions in which glial activation occurred correlated well with those that showed KA- or KA/CPT-induced neurodegeneration.
The expression of c-Jun participates in neuronal cell death (Bossy-Wetzel et al., 1997). After KA-injection, numerous pyramidal neurons in the CA3 showed apoptosis markers (Pollard et al., 1994). In this study, c-Jun phosphorylation may have preceded neurodegeneration. c-Jun phosphorylation occurred in the same region as where neurons underwent apoptotic cell death. Recently, it has been shown that the phosphorylation of c-Jun by the activation of c-Jun N-terminal kinase (JNK) is important for neuronal apoptosis using cultured cells in vitro (Bossy-Wetzel et al., 1997; Eilers et al., 1998; Herdegen et al., 1998; Schwarzschild et al., 1997; Watson et al., 1998). These reports suggest that phosphorylation of c-Jun by the activation of JNK is closely associated with KA-induced neuronal apoptosis in the rat hippocampus in vivo. In this study, we showed that treatment with CPT induces neuronal and glial changes, such as neuronal cell loss, neuronal apoptosis, and the induction of MHC class II antigen, in the CA1 as well as in the CA3. These results clearly indicate that adenosine has neuroprotective effects through similar events in the CA1 following treatment with KA.
Adenosine increased membrane potassium conductance and opened specific chloride channels through the activation of A1 receptor (Trussell and Jackson, 1985; Mager et al., 1990). Both act together in generating hyperpolarizing currents that stabilized the neuronal resting membrane potential and antagonized depolarization, and then the synaptically evoked neuronal calcium influx through voltage-sensitive ion channels is reduced (Schubert, 1988). An A1 adenosine receptor can be activated by nanomolar concentrations of extracellular adenosine present under physiological conditions in the normal rat brain (Ballarin et al., 1991). An increase in the extracellular adenosine concentration up to the micromolar range, as observed in the ischemic brain (Hagberg et al., 1987), will presumably further strengthen the A1 receptor activation and raise the threshold effect for a toxic calcium influx to cause neuronal damage. Such a protective threshold effect of endogenous adenosine against neuronal damage is supported by in vivo experiments (Donaghy and Scholfield, 1994). The massive calcium influx, through a series of as yet unclear steps that may involve phospholipase activity and the production of free radicals from the arachidonic acid pathway, eventually leads to lipid peroxidation and cell death. Based upon the concept that adenosine has neuroprotective effects (Schubert et al., 1997), the effects of adenosine receptor agonists were examined. Previously, administrated (not endogenous) adenosine and its derivatives have been shown to have neuroprotective effects against ischemia and KA-induced neurodegeneration (Heron et al., 1994; MacGregor and Stone, 1993, respectively). In this study, we found that antagonism of the A1 adenosine receptor exacerbated neurodegeneration. Furthermore, this exacerbated neurodegeneration was attenuated by the coadministration of an A1 adenosine receptor agonist. These results strongly suggest that endogenous adenosine has neuroprotective effects in neurons.
The selective vulnerability of CA3 pyramidal neurons to KA stimulation can be explained by the predominant distribution of KA receptors in the CA3 (Monaghan and Cotman, 1982; Werner et al., 1991). This hyperexcitability in the CA3 appears to be limited to within the CA3, although CA1 neurons receive synaptic inputs directly from CA3 neurons via the Schaffer collaterals. One possible pathway for CA1 neuronal death observed in this study was that CPT antagonized the protective effects of endogenous adenosine released in CA1. The synaptic transmission of the Schaffer collateral/commissural afferent in CA1 is suppressed by extracellular adenosine released during a tetanic stimulation (Sekino and Koyama, 1992; Mitchell et al., 1993; Manzoni et al., 1994). Thus, the hyperexcitation of CA3 neurons induced by the KA treatment can release adenosine that might prevents neurons in CA1 from glutamate excitotoxicity. However, the inhibitory effect of CPT against NMDA- and hypoxia-induced suppression of synaptic responses evoked by CA1 stimulus was limited to partial (Manzoni et al., 1994; Arlinghaus and Lee, 1996). These reports suggest that another additional factor can be involved in the widespread of neuronal death in CA1. Recently, a novel intrahippocampal pathway, the CA3-CA2-CA1 circuit, was identified by a physiological technique (Sekino et al., 1997). In the transverse hippocampal slices, neuronal activity in CA3 propagates to CA1 only when the slices were treated by CPT and activation of the CA3-CA2-CA1 circuit is supposed to be involved (Sekino and Obata, 1995). As seen in the present study, CPT seems to open a gate for synaptic transmission between CA3 and CA1. This study also suggests that endogenous adenosine plays a role in regulating the signal flow from CA3 to CA1. Immunohistochemical studies using an anti-A1 adenosine receptor antiserum raised against purified rat brain A1 adenosine receptor (Nakata, 1993) have shown that the adenosine A1 receptor thought to be functioning postsynaptically was distributed predominantly in the CA3a/CA2 subfield (Ochiishi et al., 1999). Removing the tonic inhibition of this field can result in enhancement of neuronal activity in CA1. The present result also supports the hypothesis that the CA2 field acts as a gate for signal flow from the CA3 to the CA1. Based on these observations, we consider that the administration of A1 adenosine receptor antagonist exacerbates neurodegeneration by i) negating the neuroprotective effects of adenosine, and reducing the threshold for bursting; and ii) reducing the threshold of CA2 neurons which contribute to the CA3-CA2-CA1 circuit.
We found that the combination of KA with an A1 adenosine receptor antagonist, CPT, induced significant neuronal cell loss in CA1 pyramidal neurons as well as in the CA3 subfield 4 days after i.c.v. injection. The activation of glial cells, such as morphological changes and the induction of MHC class II antigen, was limited to the CA3 subfield after the injection of KA alone. In contrast, treatment with KA/CPT also induced glial activation in the CA1 subfield. Although phosphorylated c-Jun, a critical marker of neuronal apoptosis, was not detected in vehicle-injected rat hippocampus, KA- and KA/CPT-injection induced phosphorylated c-Jun either in only the CA3 or in both the CA1 and the CA3, respectively. These results suggest that treatment with KA/CPT induced neurodegeneration in the CA1 through a mechanism similar to that of KA-induced neurodegeneration in the CA3. Coadministration of a specific agonist for A1 adenosine receptor, CHA, markedly attenuated the neuronal cell loss and glial activation. These results strongly suggest that endogenous adenosine has neuroprotective effects in pyramidal neurons of the hippocampus.
Jump to Author´s Recent Publications
Akiyama et al. (1988) Brain Res., 635, 257-268.
Arlinghaus & Lee (1996) Brain Res., 724, 265-268.
Ballarin et al. (1991) Acta Physiol. Scand., 142, 97-103.
Barraco et al. (1984) Neurosci. Lett., 46, 317-322.
Bloom & Fawcett (1975) In Bloom & Fawcett (eds), A text book of histology. W.B. Saunders Company, Philadelphia. pp. 1-34.
Bossy-Wetzel et al. (1997) EMBO J., 16, 1695-1709.
Coyle & Puttfarcken (1993) Science, 262, 689-695.
de Mendonca & Ribeiro (1994) Neuroscience, 62, 385-390.
Dickson et al. (1993) Glia, 7, 75-83.
Donaghy & Scholfield (1994) Brain Res., 656, 174-176.
Dragunow et al. (1985) Epilepsia, 26, 480-487.
Dunwiddie & Fredholm (1989) J. Pharmacol. Exp. Ther., 249, 31-37.
During & Spencer (1992) Ann. Neurol., 32, 618-624.
Dykens et al. (1987) J. Neurochem., 49, 1222-1228.
Eilers et al. (1998) J. Neurosci., 18, 1713-1724.
Finsen et al. (1993) Glia, 7, 41-49.
Fredholm et al. (1994) Pharmacol. Rev., 46, 143-156.
Glass et al. (1996) Brain Res., 710, 56-68.
Globus et al. (1988) J. Neurochem., 51, 1455-1464.
Hagberg et al. (1987) J. Neurochem., 49, 227-231.
Herdegen et al. (1998) J. Neurosci., 18, 5124-5135.
Heron et al. (1994) Brain Res., 641, 217-224.
Hollins & Stone (1980) Br. J. Pharmacol., 69, 107-112.
Kirino (1982) Brain Res., 239, 57-69.
MacGregor & Stone (1993) Br. J. Pharmacol., 109, 316-321.
Mager et al. (1990) Brain Res., 532, 58-62.
Manzoni et al. (1994) Science, 265, 2098-2101.
Matesic & Lin (1994) J. Neurochem., 63, 1012-1020.
Matsuoka et al. (1995) Neuroreport, 6, 2205-2208.
Matsuoka et al. (1997a) Neurochem. Int., 30, 533-542.
Matsuoka et al. (1997b) Exp. Neurol., 146, 57-66.
Matsuoka et al. (1998a) Neuroscience, 85, 1223-1233.
Matsuoka et al. (1998b) J. Cereb. Blood Flow Metab., 18, 824-832.
Matsuoka et al. (1998c) Neurosci. Lett., 252, 119-122.
McGeer et al. (1993) Glia, 7, 84-92.
Mitchell et al. (1993) J. Neurosci., 13, 3439-3447.
Monaghan & Cotman (1982) Brain Res., 252, 91-100.
Nakata (1993) Biochim. Biophys. Acta, 1177, 93-98.
Nalder et al. (1980) J. Comp. Neurol., 192, 333-359.
Ochiishi et al. (1999) Neuroscience, 93, 955-967.
Olah & Stiles (1995) Annu. Rev. Pharmacol. Toxicol., 35, 581-606.
Phillis & Wu (1981) Prog. Neurobiol., 16, 187-239.
Pollard et al. (1994) Neuroscience, 63, 7-18.
Ribeiro & Sebastiao (1984) Br. J. Pharmacol., 83, 485-492.
Schubert (1988) Brain Res., 458, 162-165.
Schubert et al. (1997) Ann. N.Y. Acad. Sci., 825, 1-10.
Schwarzschild et al. (1997) Neurosci. Lett., 148, 109-113.
Sekino & Obata (1995) Soc. Neurosci. Abstr., 21, 1101.
Sekino et al. (1997) J. Neurophysiol., 78, 1662-1668.
Taniguchi et al. (1994) Brain Res., 640, 119-125.
Trussell & Jackson (1985) Proc. Natl. Acad. Sci. USA, 82, 4857-4861.
Vallee et al. (1984) J. Cell Biol., 99, 38s-44s.
Watson et al. (1998) J. Neurosci., 18, 751-762.
Werner et al. (1991) Nature, 351, 742-744.
Young & Dragunow (1995) Exp. Neurol., 133, 125-137.
RECENT PUBLICATIONS:
Selected from 30 publications in 1994-1999.
|
|
|
|
|
|
[Cell Biology & Cytology] |
[Neuroscience] |