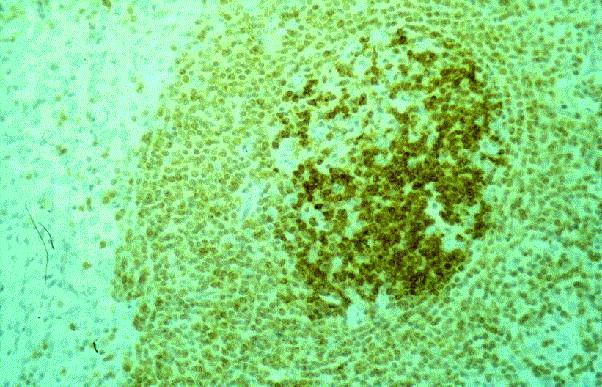
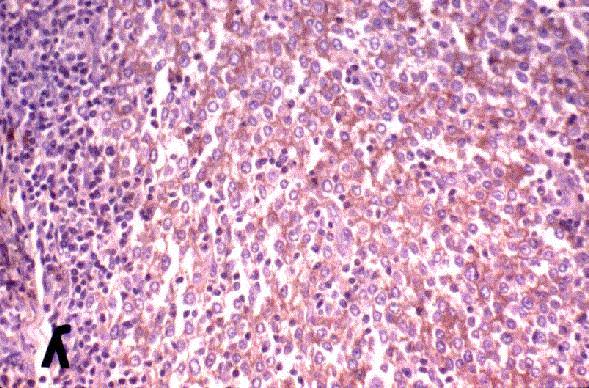
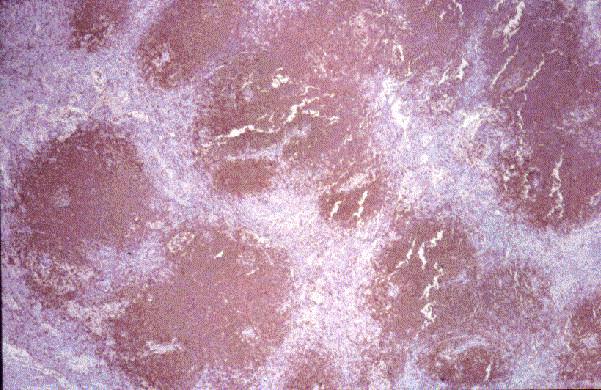
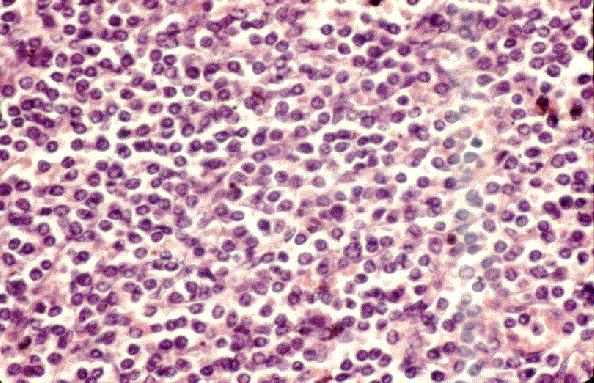
Dr. Tom·s ¡lvaro Naranjo, Dr. RamÛn Bosch PrÌncep, Dra. SalomÈ MartÌnez Gonz·lez, Dra. M™ Teresa SalvadÛ Usach
Tumores de cÈlulas B
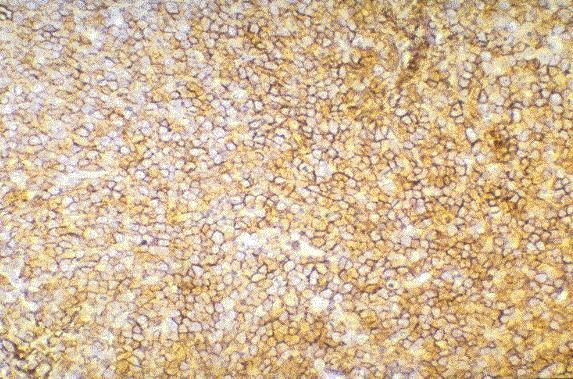
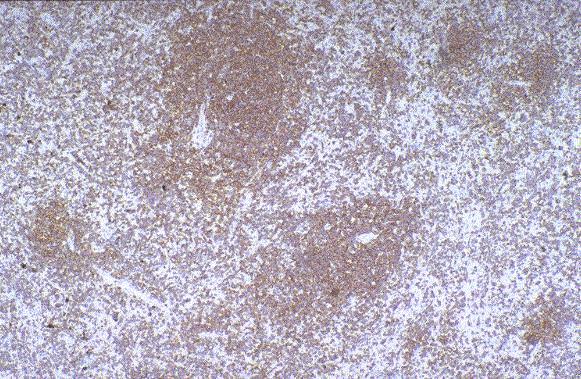
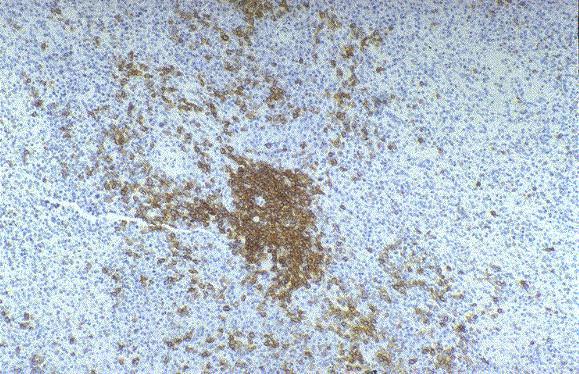
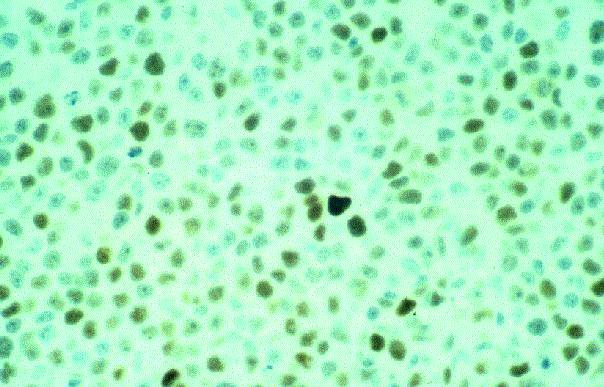
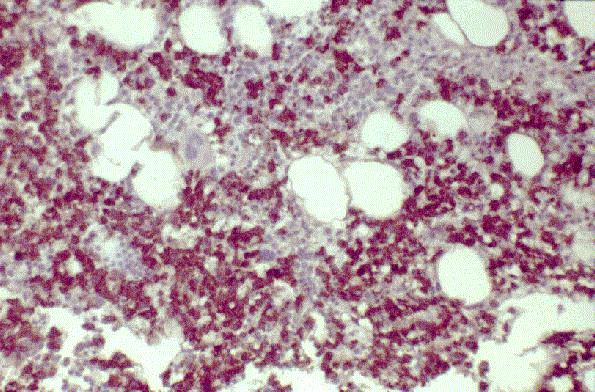
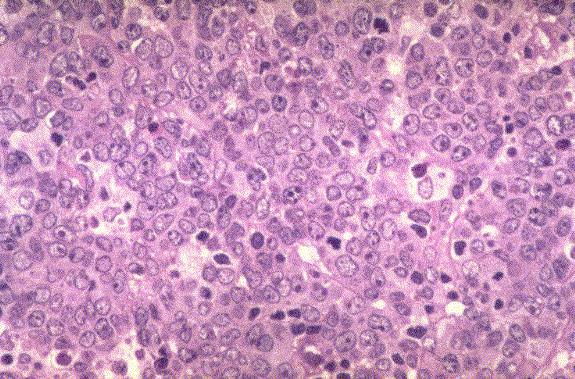
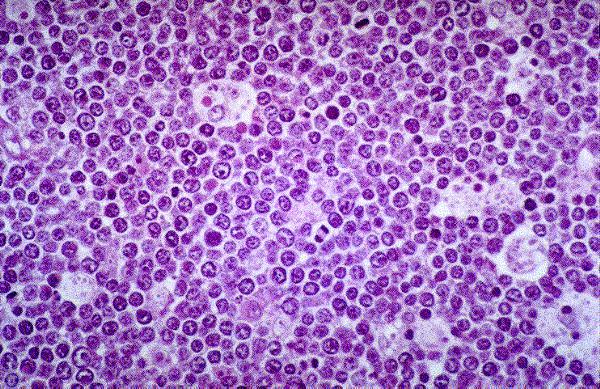
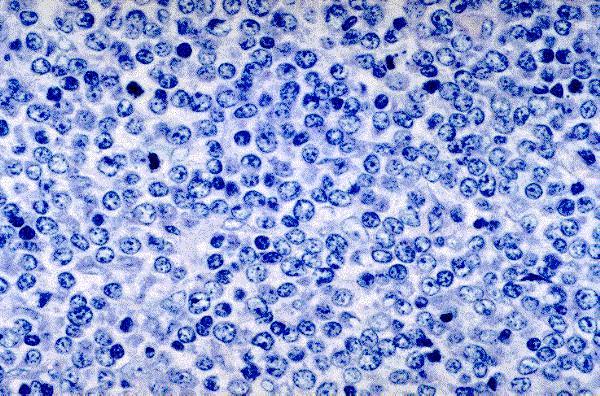
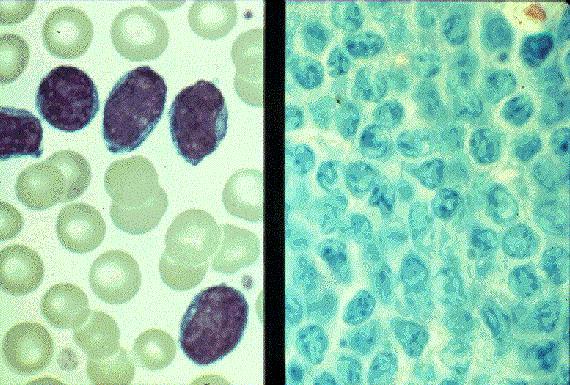
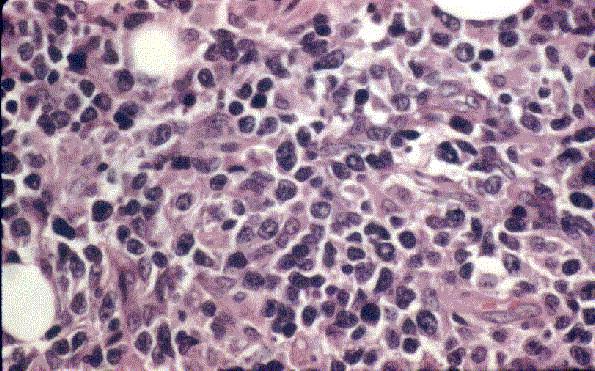
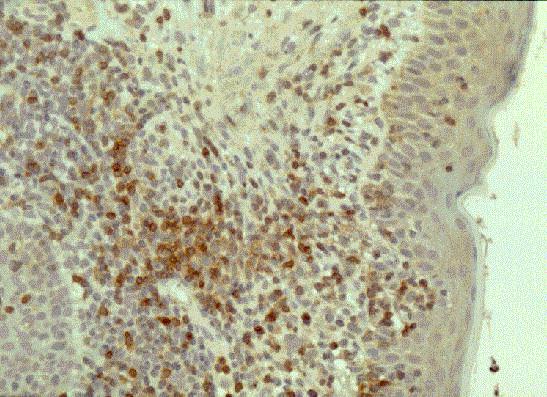
Leucemia/linfoma linfobl·stico
LEUCEMIAS / LINFOMAS LINFOBL¡STICOS B |
|
MORFOLOGÕA: |
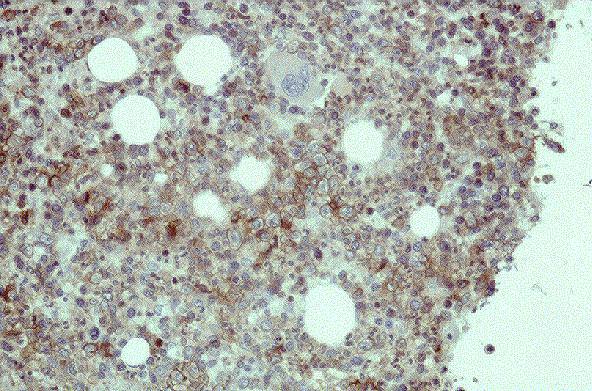
Linfocitos de tamaÒo intermedio con n˙cleo redondo o convoluto y nucleolo inconspicuo. Citoplasma escaso y dÈbilmente basÛfilo. Mitosis: Frecuentes y puede observarse un patrÛn en cielo estrellado. |
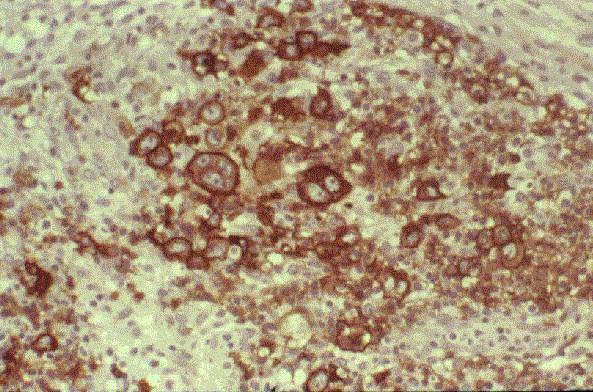
INMUNOFENOTIPO:
|
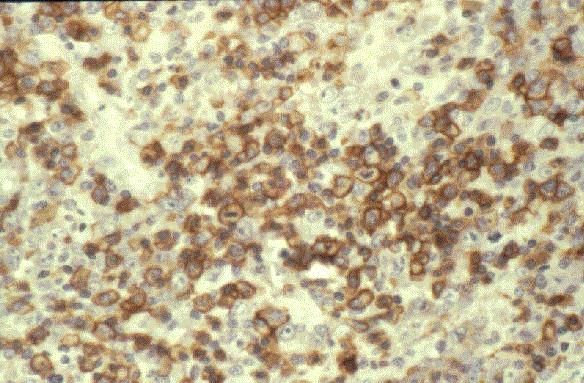
TdT+, CD19+, CD79a+, CD22+, CD20-/+, CD10+, HLADR+, cIg Mu-/+, CD34+/-. |
GEN…TICA: |
Reordena cadenas pesadas de Ig; las cadenas ligeras pueden ser reordenadas. Anomalias citogenÈticas variables. |
LEUCEMIA LINFOCÕTICA CR”NICA DE C…LULAS B LEUCEMIA PROLINFOCÕTICA DE C…LULA B LINFOMA DE LINFOCITOS PEQUE—OS B |
|
MORFOLOGÕA: |
|
INMUNOFENOTIPO:
|
|
GEN…TICA: |
|
INMUNOCITOMA / LINFOMA PLASMACÕTICO |
|
MORFOLOGÕA: |
Linfocitos plasmocitoides (citoplasma amplio basÛfilo y n˙cleo similar a linfocitos), cÈlulas plasm·ticas y linfocitos pequeÒos. Ausencia de hallazgos de LLC-B, LCM, LCF y LZM. |
INMUNOFENOTIPO:
|
SIgM +, CIg +, CD5 -, CD10 -, CD19,20,22,79a +, CD23+. |
GEN…TICA: |
No anomalÌas especÌficas conocidas. Reordenamiento de genes de IgH y IgL. |
LINFOMA DE C…LULAS DEL MANTO |
|
MORFOLOGÕA: |
Linfocitos pequeÒos irregulares de forma centrocitoide. |
INMUNOFENOTIPO:
|
Igs + , CD5 +, CD10 -/+, CD19,20,22,79a +, CD23 -. |
GEN…TICA: |
t(11;14). Reordenamiento para bcl-1. SobreexpresiÛn de PRAD1/D1 Ciclina. Genes de IgH y IgL reordenados. |
LINFOMA CENTROFOLICULAR |
|
MORFOLOGÕA: |
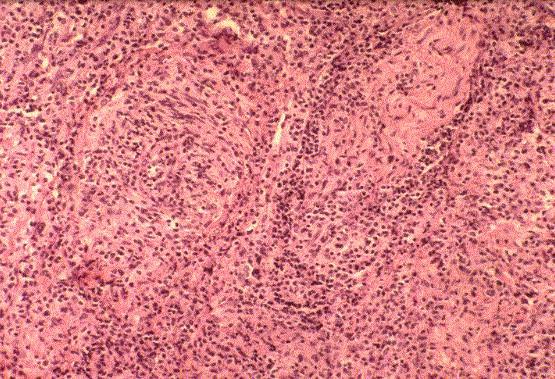
Mezcla de centrocitos y centroblastos (cÈlulas hendidas y blastos del centro germinal). PatrÛn folicular aunque puede tener ·reas difusas. Los cc predominan aunque existen los LF con predominio de CB. Grados citolÛgicos: GI: predominan las cÈl. pequeÒas. GII: mixto con cÈl. pequeÒas y grandes. GIII: predominan las cÈl. grandes. Subtipo provisional: Linfoma centrofolicular, difuso: compuesto predominantemente de cc y con menos cb pero con patrÛn difuso. |
INMUNOFENOTIPO: |
SIg + , CD5 -, CD10 -/+, CD19,20,22,43,79a +, bcl-2 +. |
GEN…TICA: |
t(14;18). Reordenamiento para bcl-2 (90% de los casos). SobrexepresiÛn de proteÌna BCL-2. Genes de IgH y IgL reordenados. |
LINFOMA DE C…LULAS B DE LA ZONA MARGINAL NODAL Y EXTRANODAL (TIPO MALT) |
|
MORFOLOGÕA: |
CÈlulas pequeÒas tipo centrocito, cÈlulas B monocitoides, linfocitos pequeÒos y cÈlulas plasm·ticas. |
INMUNOFENOTIPO: |
SIg + , CD5 -, CD10 -, CD19,20,22,79a +, CD23 -. |
GEN…TICA: |
TrisomÌa 3 y t(11;18) en algunos extranodales. No reordenamiento de bcl-2 y/o bcl-1. Genes de IgH y L reordenados. |
LINFOMA DE C…LULAS B DE LA ZONA MARGINAL ESPL…NICA |
|
MORFOLOGÕA: |
CÈlulas pequeÒas tipo centrocito, celulas B monocitoides, linfocitos y cÈlulas plasm·ticas. Centros germinales esplÈnicos hiperpl·sicos o atrÛficos pero rodeados por estas cÈl. tumorales que afectan al manto y a la zona marginal. La pulpa roja puede estar afecta. |
INMUNOFENOTIPO: |
SIg (M>G>A) +, SIg D -, CD5 -, CD10 -, CD19,20,22,79a +, CD23 -. |
GEN…TICA: |
No suficientemente estudiadas las alteraciones aunque la trisomia 3 no ha sido detectada. |
LEUCEMIA DE C…LULAS PELUDAS |
|
MORFOLOGÕA: |
Linfocitos pequeÒos con n˙cleo oval o ariÒonado y citoplasma claro. Afecta a la pulpa roja esplÈnica atrofiando la blanca. |
INMUNOFENOTIPO: |
SIg + , CD5 -, CD10 -, CD19,20,22,79a +, CD23 -, CD11c,25. CD103 (MLA) + (este es el m·s especÌfico). |
GEN…TICA: |
Ninguna especÌfica. Genes de IgH y IgP reordenados. |
PLASMOCITOMA / MIELOMA DE C…LULAS PLASM¡TICAS |
|
MORFOLOGÕA: |
CÈlulas plasm·ticas maduras o inmaduras (plasmablastos). |
INMUNOFENOTIPO: |
SIg - , CIg +, CD 38 +, CD5 -, CD10 -, CD19,20,22 -, CD79a (mb-1) +/-, CD45-/+, EMA +/-. CD30 + (BerH2 en parafina). |
GEN…TICA: |
Puede o no haber reordenamiento para IgH o IgL. |
LINFOMA DIFUSO DE C…LULAS GRANDES B |
|
MORFOLOGÕA: |
CÈlulas grandes con n˙cleo vesicular de gran tamaÒo (de almenos dos veces el tamaÒo de un linfocito pequeÒo), nucleolo prominente y con citoplasma basÛfilo on un Ìndice de proliferaciÛn moderado-alto. Esta categoria incluye los linfomas centroblasticos (a veces multilobados), inmunobl·ticos, anapl·sicos B (CD30+ o no) y los Linfomas B ricos en cÈlulas T o ricos en histiocitos. Subtipo: Linfoma primario mediastÌnico (tÌmico) de cÈlulas grandes B: centroblastos, centrocitos o cÈl. multilobadas con citoplasma claro. Raramente parecen inmunoblastos. Frecuente esclerosis compatimentalizante. Suele afectar el timo en la presentaciÛn. |
INMUNOFENOTIPO: |
CIg +/-, SIg -/+, CD5 -/+, CD10 -/+, CD19,20,22,79a +, CD45 +/-. |
GEN…TICA: |
t(14;18) y reordenamiento de bcl-2 en un 30% y c-myc en algunos casos. . Reordenamiento de bcl-6 hasta un 60%. Reordenamiento de genes de IgH y IgL |
LINFOMA DE BURKITT |
|
MORFOLOGÕA: |
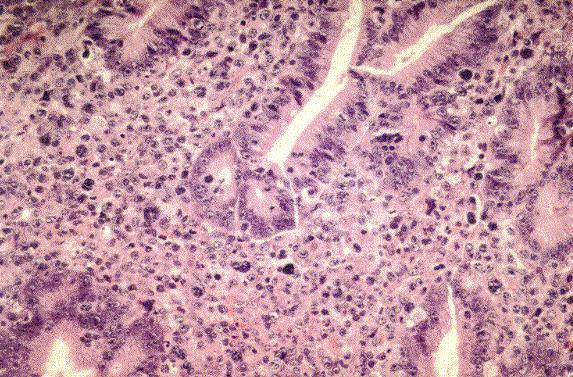
CÈlulas monomÛrficas, de mediano tamaÒo, con citoplasma basofÌlico amplio, n˙cleo redondo, nucleolos m˙ltiples y con altÌsimo Ìndice de mitosis y cariorexis y apariencia de cielo estrellado. PatrÛn celular cohesivo. |
INMUNOFENOTIPO: |
SIgM + , CD5 -, CD10 +, CD19,20,22,79a +, CD23 -. |
GEN…TICA: |
t(8;14), t(2;8), o t(8;22). Reordenamiento del c-myc. EBV en la mayorÌa de los casos africanos y en un 25-40% de los asocidados a inmunodeficiencias y menos en el resto. |
LINFOMA B DE ALTO GRADO, BURKITT-LIKE Entidad provisional |
|
MORFOLOGÕA: |
Hallazgos intermedios entre los LCGB de tipo centroblastico y inmunobl·stico y los Linfomas de Burkitt y las subclasificaciÛn en uno de estos grupos es imposible. Es una categorÌa difÌcilmente reproducible y heterogÈnea pero necesaria para estos casos borderline. |
INMUNOFENOTIPO: |
SIg +/- (pueden tener CIg) , CD5 -, CD10 -, CD19,20,22,79a +. |
GEN…TICA: |
Carecen de reordenamiento del c-myc. Reordenamiento para bcl2-2 (30%). |
Volver al Õndice
Tumores de cÈlulas T y cÈlulas NK
Leucemia/linfoma linfobl·stico
LEUCEMIA / LINFOMA LINFOBLASTICOS T |
|
MORFOLOGÕA: |
CÈlulas tumorales idÈnticas a los linfoblastos B con n˙cleo redondo o convoluto, cromatina fina, nucleolo inconspicuo y citoplasma escaso. |
INMUNOFENOTIPO: |
TdT +, CD1a +/-, CD3 +/,CD7 +. CD4 y CD8 doble positivo o negativo. Marcadores B negativos. Ocasionalmente expresan marcadores de NK (CD 16, CD57). |
GEN…TICA: |
Reordenamiento de los genes TCR variable. TambiÈn reordenamientos de IgH. AnomalÌas citogenÈticas variables. |
LEUCEMIA LINFOCÕTICA CR”NICA T LEUCEMIA PROLINFOCÕTICA T |
|
MORFOLOGÕA: |
La mayorÌa linfocitos con nucleolo prominente, con alguna irregularidad nuclear y con citoplasma m·s abundante que las LLC-B perteneciendo a la categorÌa prolinfocÌtica. Algunos casos tienen cÈlulas m·s pequeÒas y parecen la LLC-B. AfectaciÛn difusa y paracortical. Carece de pseudofolÌculos. Las vÈnulas de endotelio alto pueden ser prominentes. La pulpa roja esplÈnica y los sinusoides hepatocitarios pueden estar infiltrados. La mÈdula suele estar infiltrada difusamente. |
INMUNOFENOTIPO: |
CD2,3,5,7 +, CD4 + (65%), CD8 +/ -, CD25 -. |
GEN…TICA: |
Reordenamiento clonal para genes TCR. Inv 14(q11;32) en un 75%. Trisomia 8q. |
LEUCEMIA DE LINFOCITOS GRANULARES GRANDES: tipo cÈlulas T tipo cÈlulas NK |
|
MORFOLOGÕA: |
CÈlulas de mediano tamaÒo a pequeÒo con n˙cleo redondo u oval, excÈntrico. Gr·nulos azurÛfilos citopl·smicos. |
INMUNOFENOTIPO: |
Tipo CÈl. T: CD2+, CD3+, CD5-, CD7-, TCRab+, CD4-, CD8+, CD16+ CD56- CD57+/-, CD25-. Tipo CÈl. NK: CD2+, CD3-, TCRab-, CD4-, CD8+/-, CD16+ CD56+/- CD57+/-. |
GEN…TICA: |
Tipo CÈl. T: Reordenamiento de genes TCR clonal. Tipo CÈl. B: No reordenamiento de genes TCR clonal. AsociaciÛn con el VEB en casos agresivos asi·ticos. No anomalÌas especÌficas. |
MICOSIS FUNGOIDES / SÕNDROME DE S…ZARY |
|
MORFOLOGÕA: |
CÈlulas de mediano a pequeÒo tamaÒo, con n˙cleo cerebriforme. MinorÌa de cÈlulas mayores con idÈntica morfologÌa nuclear. InfiltraciÛn epidÈrmica. AfectaciÛn paracortical con acompaÒamiento por cÈlulas dendrÌticas y CÈl. De Langerhans. |
INMUNOFENOTIPO:
|
CD2,3,5 +, CD4+ en la mayorÌa, CD7+ en 1/3, CD8,25 -. |
GEN…TICA: |
Reordenamientos clonales para genes TCR. No anomalÌas especÌficas. |
LINFOMA DE C…LULAS T PERIF…RICAS, NO ESPECÕFICO |
|
MORFOLOGÕA: |
Mezcla de CÈl. T de pequeÒo y gran tamaÒo, con n˙cleo cerebriforme. Incluso en los que predomina una poblaciÛn intermedia o grande existe un amplio espectro de tamaÒos celulares. Los eosinÛfilos, histiocitos epitelioides y vasos pueden ser numerosos (El linfoma de Lennert o linfoma de cÈlulas linfoepiteliodes se considera una categorÌa citolÛgica de LT perifÈrico rica en histiocitos epiteliodes). Ocasionales cÈlulas hipercrom·ticas que pueden simular CRS. InfiltraciÛn epidÈrmica. Tres categorÌas provisionales citolÛgicas: De cÈlula mediana-pequeÒa. Mixtos. De cÈlula grande. Cuatro tipos de L. T perifÈrico "especÌficos": Linfoma T angioinmunobl·stico. Linfoma angiocÈntrico. L T intestinal con o sin enteropatÌa. Linfoma/leucÈmia de cÈlulas T del adulto (HTLV1+) Dos entidades provisionales: Linfoma hepatoesplÈnico de cÈlulas Tg d . Linfoma paniculÌtico subcut·neo de cÈlulas T. |
INMUNOFENOTIPO: |
ExpresiÛn variable de marcadores T: CD3,2,5,7; El DC3 es el m·s estable. |
GEN…TICA: |
Reordenamiento clonal de genes de TCR. No anomalÌas especÌficas. |
LINFOMA DE C…LULAS T ANGIOINMUNOBL¡STICO |
|
MORFOLOGÕA: |
Arquitectura del ganglio borrada, senos perifÈricos abiertos i incluso dilatados aunque el infiltrado se extiende m·s all· de la c·psula. ProliferaciÛn de vÈnulas de endotelio alto arboriz·ndose muchas mostrando paredes engrosadas o hialinizadas PAS+. Agregados de CDF que rodean los vasos sanguÌneos proliferantes y que pueden dar la apariencia de centros germinales quemados. Ausencia de folÌculos secundarios. Infiltrado mixto de linfocitos pequeÒos, inmunoblastos y cÈlulas atÌpicas claras. |
INMUNOFENOTIPO: |
CD2,3,5,7 +. Generalmente CD4 +. Grandes agrupaciones de cÈlulas dendrÌticas foliculares alrededor de las vÈnulas que proliferan. Ocasionalmente blastos B. |
GEN…TICA: |
Reordenamiento clonal de genes par TCR en 75%. IgH en 10%. Genoma de VEB en muchos casos. TrisomÌa 3 y/o 5 puede ocurrir. |
LINFOMA ANGIOC…NTRICO |
|
MORFOLOGÕA: |
Linfocitos polimorfos que rodean e infiltran la pared de los vasos compuestos por una mezcla de linfocitos pequeÒos de apariencia normal y un n˙mero variable de linfocitos atÌpicos y inmunoblastos junto con cÈlulas plasm·ticas y ocasionales eosinÛfilos y histiocitos. Provocan oclusiÛn de la luz vascular y por ello se acompaÒan de abundante necrosis de tejido tumoral y normal. La granulomatosis linfomatoidea que afecta a los pulmones ha sido considerada en este grupo pero recientemente se ha sugerido que al menos una parte de estos casos pueden ser proliferaciones de cÈlulas B asociadas a VEB y por tanto una categorÌa diferente. |
INMUNOFENOTIPO: |
CD2+, generalmente CD56+ y CD3 -. CD5,7 +/-, CD4,8 +/-. En casos pulmonares las cÈlulas atÌpicas expresan antÌgenos B. |
GEN…TICA: |
Reordenamiento clonal de genes TCRb. |
LINFOMA DE C…LULAS T INTESTINAL (con o sin enteropatÌa) |
|
MORFOLOGÕA: |
⁄lceras yeyunales, generalmente m˙ltiples y con perforaciÛn frecuente que se acompaÒan o no de masa. El infiltrado es polimorfo con cÈl. PequeÒas o medianas mezcladas con cÈl. Grandes o anapl·sicas generalmente acompaÒ·ndose de una importante cantidad de cÈl. T intraepiteliales. La mucosa no neopl·sica puede o no mostrar atrofia vellositaria. Las lesiones iniciales pueden mostrar ˙nicamente aisladas cÈl. AtÌpicas y numerosos histiocitos reactivos. |
INMUNOFENOTIPO: |
CD3,7 +, CD8 +/ -, CD103 + (MLA: HML-1, LFG1, Bly7, Ber-ACT8). |
GEN…TICA: |
Reordenamiento clonal de genes de TCRb. |
LINFOMA / LEUCEMIA DE C…LULAS T DEL ADULTO |
|
MORFOLOGÕA: |
Infiltrado con patrÛn difuso linfocitos atÌpicos polimorfos de tamaÒo pequeÒo que se mezclan con linfocitos de gran tamaÒo, todos ellos con n˙cleos pleomÛrficos. Pueden observarse cÈlulas gigantes que parecen CRS. En sangre perifÈrica se observan frecuentemente cÈl. con n˙cleos hiperlobados (cÈlulas en flor o en hojas de trÈbol). La infiltraciÛn de la mÈdula es difusa. |
INMUNOFENOTIPO: |
CD2,3+, frecuentemente CD4,25 +, CD7 -. |
GEN…TICA: |
Reordenamiento clonal de los genes de los TCR. Genoma del HTLV-1 integrado en todos los casos. |
LINFOMA DE C…LULAS GRANDES ANAPL¡SICO (CD30+) (tipos T y de CÈl. Nula). |
|
MORFOLOGÕA: |
CÈlulas bl·sticas, de gran tamaÒo y bizarras con n˙cleos pleomÛrficos, con forma de herradura o con multinucleaciÛn generalmente con uno o m˙ltiples nucleolos prominentes. Las formas multinucleadas simulan CRS. Citoplasma abundante. PatrÛn cohesivo con extensiÛn intrasinusoidal. Frecuente afectaciÛn extranodal (partes blandas, hueso, piel). Mezcla de granulocitos y macrÛfagos. |
INMUNOFENOTIPO: |
Fenotipo T o nulo. CD 30 +, EMA +/-. CD45 +/-, CD25+/-, CD15 -/+, CD3 -/+, CD43 -/+, CD45RO -/+, CD68 - (KP1 puede ser +), lisozima -. Los casos cut·neos son EMA - y antÌgeno de los linfocitos cut·neos (CLA, HECA-452). |
GEN…TICA: |
t(2;5) en algunos casos de formas sistÈmicas. Reordenamiento para los genes TCR en 50-60% de los casos. |
LINFOMA ANAPL¡SICO DE C…LULAS GRANDES RELACIONADO CON HODGKIN - Entidad provisional - |
|
MORFOLOGÕA: |
Nidos confluentes de cÈlulas tumorales con patrÛn cohesivo, generalmente sinusoidal, similar al LACG pero con hallazgos arquitecturales que asemejan EH EN tale como un engrosamiento capsular, un crecimiento nodular del tumor y bandas de esclerosis. |
INMUNOFENOTIPO: |
Fenotipo T o nulo. CD 30 +, EMA +/-. CD45 +/-, CD25+/-, CD15 -/+, CD3 -/+, CD43 -/+, CD45RO -/+, CD68 - (KP1 puede ser +), lisozima -. EBV -. |
GEN…TICA: |
No conocidas. |
Volver al Õndice
[Õndice] [Marcadores] [Linfomas_NH] [Enf._Hodgkin] [Prolif._histiocitarias] [Sd. linfoprolif._esplenicos] [Extranodales] [Dg._pr·ctico] [BibliografÌa] [IconografÌa] |