|
Información || Congresos || Cursos || Territoriales || Noticias || Patología || Telepatología |
|
Información || Congresos || Cursos || Territoriales || Noticias || Patología || Telepatología |
| . |
Reunión de la Asociación Territorial de
Madrid
Dpto. de Anatomía Patológica. ATROFIA ESPINAL MUSCULAR INFANTIL TIPO 0 Se ha descrito recientemente un nuevo tipo de atrofia espinal muscular infantil (enfermedad de Werdnig-Hoffman), llamado tipo 0. Presentamos una observación de esta variedad de enfermedad de Werdnig-Hoffman que se caracteriza por la gran intensidad de sus manifestaciones clínicas, la mayor extensión de sus lesiones neurológicas y por tener un comienzo prenatal. CASO CLÍNICO: Tras 41 semanas de gestación nace un niño cuya madre, de 36 años, era primípara y primigesta. En el embarazo hubo polihidramnios y se realizó amniocentesis con resultado de cariotipo normal, 46 XY. Desde el nacimiento fue necesaria la ventilación mecánica por ausencia de esfuerzo respiratorio. La hipotonía era generalizada y severa con los rasgos dismórficos propios. En las radiografías, los huesos largos y las costillas eran gráciles y había una fractura de tercio distal de húmero izquierdo. El EMG mostró signos de denervación y ausencia de conducciones motoras y sensitivas. La biopsia muscular y de nervio periférico demostró atrofia difusa de fibras musculares y disminución moderada del número de fibras mielínicas y signos de hipomielinización. El niño falleció a los 16 días de vida.
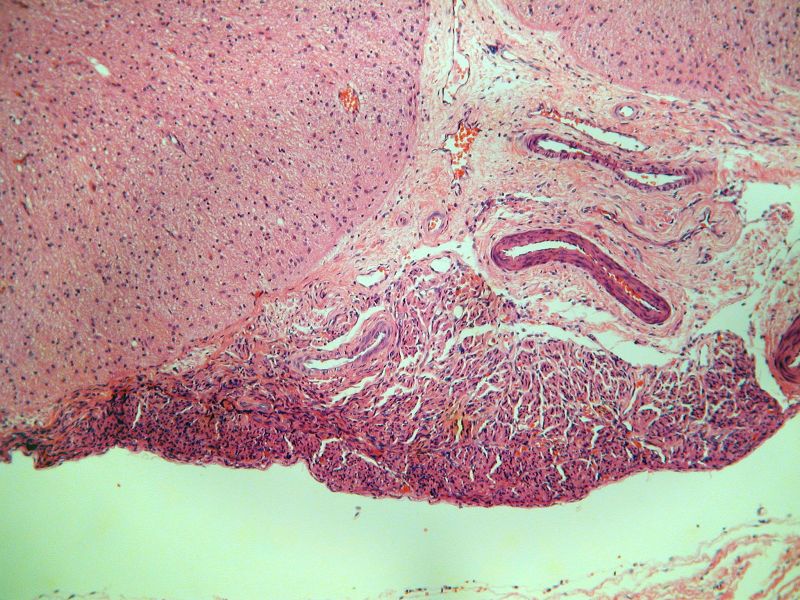
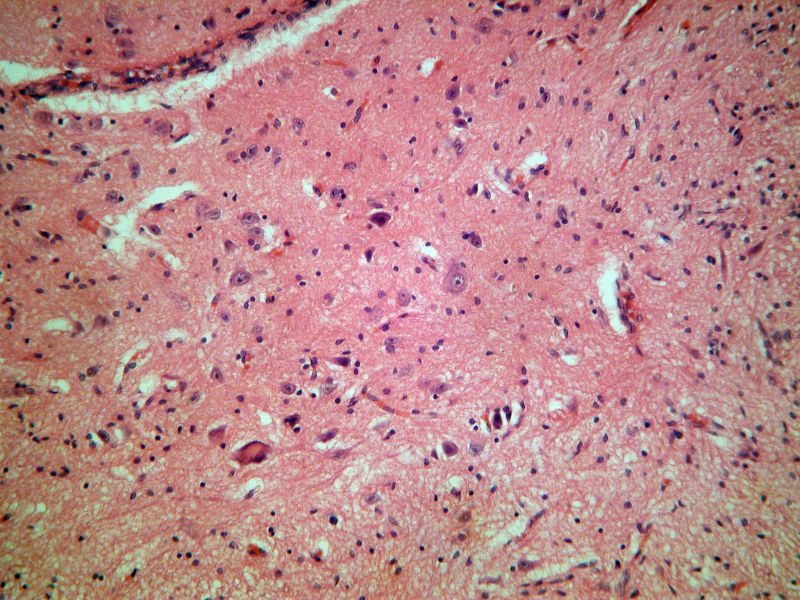
Hospital la Paz. Madrid En la autopsia, el recién nacido pesaba 3150 g. y tenía deformaciones faciales y contracturas articulares múltiples propias de la secuencia de hipocinesia fetal. El estudio radiológico postmortem mostró la existencia de otra fractura en la diáfisis del fémur derecho. En disintos músculos estriados se encontraron fibras pequeñas y redondeadas (histoquimicamente de tipo I) y otras de forma normal y tamaño aumentado. La médula espinal tenía una intensa disminución del número de motoneuronas del asta anterior acompañada de gliosis. Las motoneuronas existentes mostraban marginación de la sustancia tigroide, cromatolisis central, desplazamiento nuclear y figuras de neuronofagia. Alteraciones semejantes se encontraron en la columna de Clark, los núcleos motores del tronco del encéfalo (incluido el del III par craneal), la oliva bulbar, los núcleos propios del puente, los núcleos profundos del cerebelo, sustancia negra, múltiples núcleos talámicos, hipotálamo, núcleo subtalámico, putamen y globos pálidos. El estudio molecular mostró delección en homocigosis del gen SNMt (exones 7 y 8 analizados). COMENTARIOS: La atrofia muscular espinal infantil es una enfermedad neurodegenerativa de herencia autosómica recesiva que tradicionalmente se ha subdividido en tres tipos en base al momento de aparición y la gravedad del cuadro: el tipo I, o enfermedad de Werdnig-Hoffman, es la forma más grave y de comienzo antes de los seis meses, el tipo II es la forma intermedia y comienza entre los seis y los 18 meses y el tipo III, o enfermedad de Kugelberg-Welander, es la forma más leve y de comienzo más allá de los dieciocho meses.
Hospital la Paz. Madrid
|
. | |
| © SEAP. Sociedad Española de Anatomía Patológica | Actualizado: 24/09/2002 |
||