FIBROMA OVARICO CALCIFICADO EN LA INFANCIA.
I. Colmenero Blanco, I. de Prada Vicente, I. González Mediero.
Hospital Universitario Niño Jesús, Madrid.
HISTORIA CLíNICA
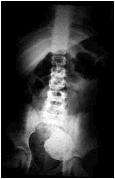
Niña de 7 años diagnosticada a los 8 meses de vida de Neuroblastoma rico en estroma estadio I en mediastino posterior, tratada con resección quirúrgica completa y libre de enfermedad hasta la actualidad. Consulta en Urgencias por dolor abdominal inespecífico. A la palpación se aprecia una masa en fosa ilíaca izquierda. Se realizan exploraciones complementarias:
Se realizó ooforectomía izquierda y biopsia del ovario derecho.
ESTUDIO ANATOMOPATOLÓGICO
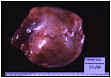
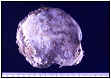
La pieza quirúrgica corresponde a una tumoración redondeada de 6 cm, blanco-grisácea, bien delimitada, de consistencia pétrea adherida a ovario de aspecto externo normal (fig.
2). A la sección del ovario se observa un nódulo de 0,4 cm con las mismas características que el anterior. En la cuña del ovario contralateral aparece otra formación con el mismo aspecto macroscópico de 0,7 cm.
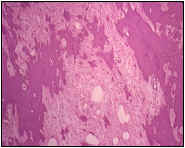
El estudio histológico de los tres nódulos (fig. 3) demuestra una proliferación de hábito mesenquimal, constituída por células fusiformes, monomorfas, sin atipias ni mitosis; que crecen formando fascículos entrelazados. La célularidad es en general escasa y con una intensa colagenización. Se observan frecuentes zonas de edema intersticial y presencia de grandes placas irregulares hialinas calcificadas. Las lesiones se hallan bien delimitadas y el tejido ovarico adyacente no muestra alteraciones significativas
DISCUSIÓN
Los tumores del estroma ovárico, entre los cuales se incluye el Fibroma, son infrecuentes en la edad pediátrica. Su presencia en este grupo, especialmente si son bilaterales y calcificados, es altamente sugestiva del Síndrome Nevoide Basocelular (Gorlin-Goltz).
El Síndrome de Gorlin (AD/esporádico) se caracteriza por la aparición progresiva durante la infancia de nevus basocelulares con potencial de transformación maligna, queratoquistes mandibulares, calcificación de la hoz cerebral y cerebelosa, hoyuelos palmoplantares, estrabismo y otras alteraciones malformativas. Se ha asociado con distintos tipos de tumores como meduloblastomas, fibromas, lipomas y neurofibromas.
En nuestra paciente los únicos rasgos fenotípicos presentes son un estrabismo convergente, hipertelorismo, macrocefalia, talla superior al percentil 95 y la presencia de Fibromas ováricos bilaterales calcificados. Dada la corta edad de la paciente cabría esperarse la aparición evolutiva de otras lesiones propias del Síndrome.
El diagnóstico definitivo se realiza mediante la identificación de mutaciones del gen PATCHED localizado en el cromosoma 9, banda q22 (pendiente de resultados). De confirmarse el diagnóstico este sería el primer caso de asociación del Síndrome de Gorlin-Goltz con Neuroblastoma.
BIBLIOGRAFIA
1. Gorlin RJ, Goltz RW: Multiple nevoid basal cell epithelioma, jaw cysts, and bifid ribs. A syndrome. N Engl J Med 262:908, 1960
2. Scully RE, Galdabini JJ, McNelly BU. Case Records of the Massachusetts General Hospital. N Engl J Med 294:14, 1976
3. Johnson AD, Herbert AA, Esterly NB. Nevoid basal cell carcinoma syndrome: bylateral ovarian fibromas in a 3 1/2-year old girl.
J Am Acad Dermatol 14:371-374, 1986
4. Compton JG, Goldstein AM, Turner M et al. Fine mapping of the locus for nevoid basal cell carcinoma syndrome on chromosome 9q.
J Invest Dermatol 103:178-181, 1994