| . |
[XXII
Congreso Nacional (Palma de Mallorca)]
[XXI
Congreso Nacional (Madrid)] [ XX Congreso Nacional
(Pamplona) ] [XIX Congreso Nacional (Barcelona) ]
[XVIII Congreso Nacional (Málaga) ]
CURSO CORTO LÍMITES DE LA PATOLOGÍA
Nefropatía crónica del trasplante (NCT)
Javier Pardo
Clínica Universitaria, Universidad de Navarra, Pamplona (Navarra).
jpardo@unav.es
Introducción
La NCT, vasculopatía del trasplante o rechazo crónico es
un deterioro progresivo de la función del injerto (en al menos dos medidas
con tres meses de intervalo, comenzando al menos 3 meses después del tx) en
ausencia de cualquier otra enfermedad y después de la confirmación del
diagnóstico por medio del estudio anatomopatológico1.
Se trata de una lesión indolente pero progresiva e irreversible, que afecta
en diferente grado a todos los trasplantes de órganos sólidos, pero con
significación clínica tiene una incidencia a los 5 años varía entre 10-20%
en hígado a 50-60% en pulmón. La NCT es la causa de más frecuente (34%) de
pérdida del injerto, después del primer año del trasplante.
Los aspectos patológicos comunes de la NCT son:
Inflamación y fibrosis, arteriolopatía obliterativa y atrofia del injerto.
Fisiopatología de la NCT
La patogenia de la NCT se resume en la figura 1
2.
El carácter inmune de la NCT se establece por su
relación con episodios de rechazo agudo3, el que
la identidad antigénica (HLA) prevenga del NCT en xenotrasplantes4,
el que la terapia anti-CD4 y anti-CD8 elimine problemas retrasplante, el que
al aumentar el tratamiento inmunosupresor se frene la progresión del NCT y
finalmente la alosensibilización previa aumenta los episodios de RA y la
supervivencia del injerto. La célula diana es el endotelio que interacciona
con el sistema del complemento, la coagulación, la respuesta inflamatoria,
los leucocitos circulantes. El sistema inmune, el músculo liso de los vasos
y la matriz extracelular que le rodea5.
En la patogenia del NCT intervienen también factores
humorales producidos por:
Se ha sugerido algunos factores no inmunológicos
del NCT basados en los siguientes argumentos:
-
Donantes vivos no relacionados o idénticos son mejores
que los donantes añosos de similares identidades.
-
Relación con infecciones principalmente virales6
y con determinados fármacos
-
Injertos con tolerancia antigénica pueden desarrollar
NCT7
-
Los retrasplantes en las mismas ratas donantes
desarrollan vasculopatía
-
La isquemia aumenta la síntesis de mediadores de la
inflamación (citoquinas, moléculas de adhesión). La isquemia en el rechazo
crónico se produce como consecuencia de una endarteritis arterial
proliferativa, que causa un daño directo a los órganos
-
Donantes añosos es posible que tengan peor pronóstico
por tratarse de riñones con reducción de la masa renal funcional
-
La hipertensión aumenta la incidencia de NCT8
- La hiperfiltración (masa renal funcionante), la arteriosclerosis/hiperlipidemia
y la existencia de glomeruloesclerosis son factores de riesgo de NCT (Fig.
1).

Si predominan los agentes inmunológicos, la fibrosis
intersticial comienza en la zona de separación corticomedular, los
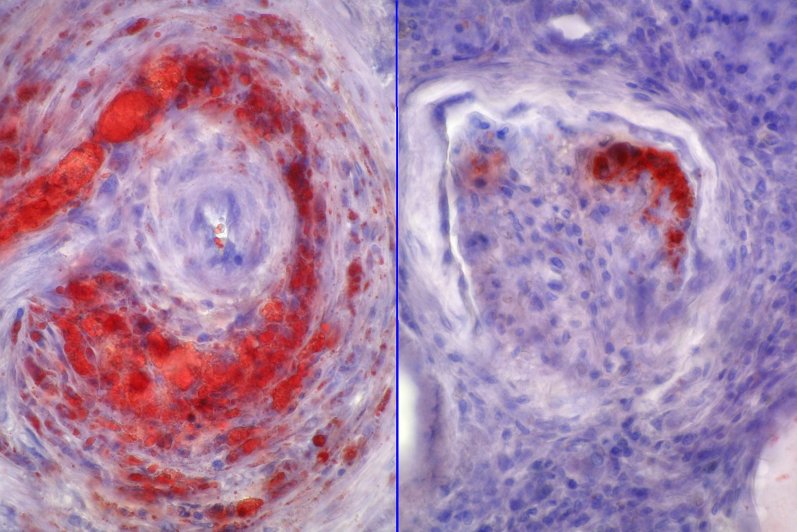
glomérulos no presentan lesiones, los vasos presenta vasculitis (Fig. 2) y
los pacientes tienen antecedentes de varios episodios de rechazo agudo. Si
son más importantes los factores no inmunes, la fibrosis comienza en región
subcapsular, aparecen lesiones de glomerulosclerosis focal (Fig. 3), los
vasos tienen lesiones de tipo arteriosclerótico y la evolución del rechazo
es muy lenta.

Uno de los mecanismos esenciales de la aparición de
lesiones del NCT precisan de la síntesis de moléculas de adhesión, que
favorezcan el intercambio molecular de las células que participan en el NCT,
principalmente endotelio, células de la inflamación, músculo liso y
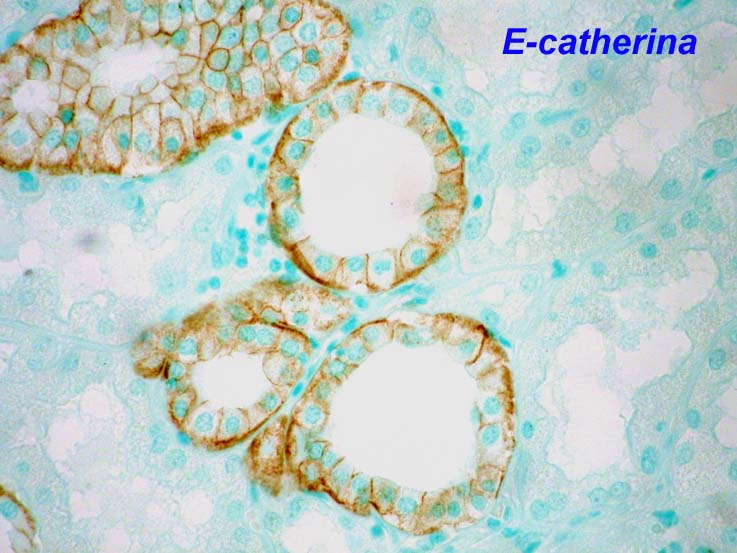
fibroblastos. Son especialmente importantes las integrinas, caterinas y
selectinas. EN los injertos aumenta la expresión de ICAM-1 y VCAM-1 en el
endotelio y epitelio tubular; en el intersticio aumentan VLA-4 y LFA-1 al
mismo tiempo que la infiltración linfoide. ICAM-1 y VCAM-1 desaparecen en el
riñón terminal9. Así el aumento de reactividad
de ICAM-1 en el endotelio acelera la aparición de lesiones arteriales del
tipo de arteriosclerosis en el injerto10.
HLA-DR y moléculas de adhesión
HLA-DR
endotelial y
Reactividad ICAM-1 |
Arteriosclerosis
No Sí |
Tiempo
libre de
Arteriosclerosis |
|
Negativo (n=17) |
16
1 |
37,4+39 meses |
|
Positivo (n=53) |
18
35 |
23,5+2,1 meses |
Por otro lado algunas moléculas de adhesión (ICAM-1 and
VCAM-1) aumentan como consecuencia de infecciones virales11.
EL aumento de algunas moléculas de adhesión como las selectinas aumenta el
riesgo de arteriosclerosis12 (Fig. 4)

La remodelación tisular del injerto se favorece por medio
del control de la apoptosis de todas las células del injerto. El número de
células en apoptosis aumenta conforme aumenta el grado de rechazo13:
Marcadores de apoptosis se encuentran en el NCT de todos
los tx.
La apoptosis puede ser la vía común de las lesiones de
los NCT
La acomodación del injerto a los anticuerpos es posible
por medio de la activación de genes anti-apoptosis.
Las lesiones vasculares que se encuentran en el NCT son
de dos tipos14.
:
1. Agudas
a. Necrotizantes
IgM, C3, fibrina
b. Infiltrativas
Infiltrados de células mononucleadas,
Células xantomatosas
2. Crónicas
a. Proliferativas
Fibroblástica. Músculo liso.
Polimorfismo, hipercromasia, alteración núcleos endotelio.
b. Esclerosantes
Fibrosis intimal.
Fragmentación elástica int.
Hialinización
Diagnóstico diferencial
El NCT hay que diferenciarlo de la toxicidad crónica por
CsA
| |
NCT |
Toxicidad por CsA |
Glomérulo
Tamaño |
Normal |
Disminuido |
| MB |
Duplicación |
Engrosada o normal |
|
Capilares |
Patente |
Trombosis o GS focal |
| IF |
IgG lineal |
IgG lineal |
|
Intersticio |
Fibrosis |
Fibrosis lineal |
| Túbulos |
Derrame tubular |
Atrofia focal |
| Vasos |
Hiperplasia intimal
Hipertrofia intimal
Estrechamiento luz |
Depósitos nodulares
hialinos en la media
y en la íntima |
Los factores que aumentan el riesgo de progresión a fallo
renal terminal son: Hipertensión, albuminuria o proteinuria, pobre control
de la glucemia, tabaco, exceso de proteínas en la dieta e hiperlipidemia.
Diagnóstico precoz de la NCT
El diagnóstico precoz podría realizarse por:
-
Expresión del MHC: Aumento de la autotolerancia
-
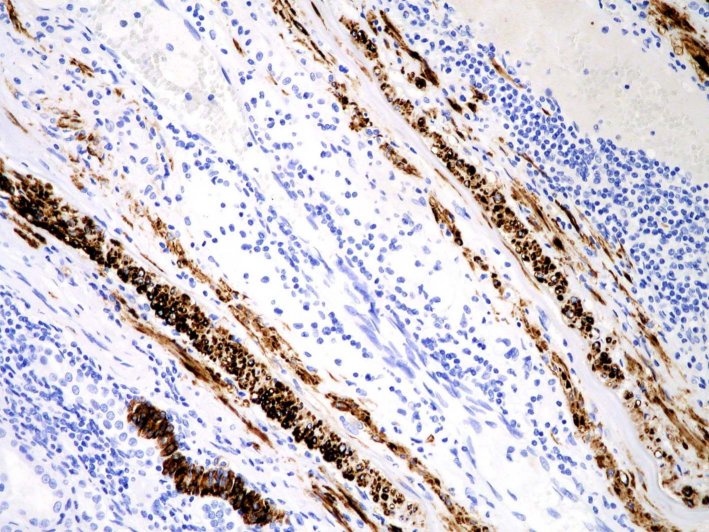
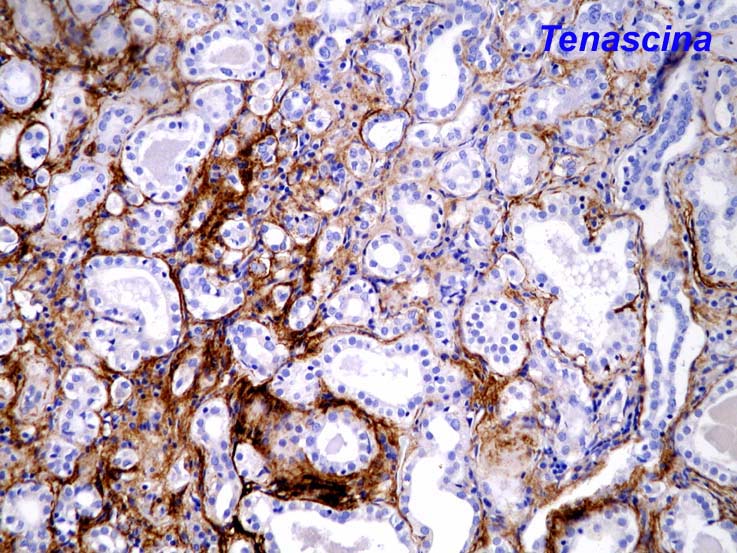
Depósito de tenascina (Fig. 5) y fibrina microvascular15
-
Deplección de activador del plasminógeno tisular
vascular (tPA)
-
Pérdida de antitrombina vascular
-
Troponina I
-
Elevación de moléculas de adhesión (VCAM-1)
-
Quimioquinas y sus receptores
- Activación de linfos T

Prevención de la NCT
Según los datos conocidos sobre la patogenia del NCT,
podría hacerse una prevención del mismo por medio de16-18
-
Mayor igualdad HLA
-
Mejor inmunosupresión
-
Disminución del tiempo de isquemia fría
-
Suero antilinfocitario?
-
Inhibición de moléculas de adhesión como LFA-1/ICAM-1
and VLA-4/VCAM-1 19. Las interacciones
endotelio/linfocitos T puede neutralizarse con anticuerpos anti-VCAM y
anti-E-selectin 20
-
Profilaxis infecciosa. CMV?
-
Profilaxis RA (donantes, compliance, mejor
inmunosupresión
)
-
Profilaxis hipertensión
-
Profilaxis hiperlipidemia
La lesión es irreversible y los tratamientos curativos
son por ahora totalmente ineficaces, salvo el retransplante.
Concluimos que:
-
VT es la principal causa de fracaso de tx a largo
plazo.
-
En su patogénesis intervienen muchos factores que
impiden encontrar un tratamiento adecuado.
-
Complicación de demostrar el efecto beneficioso de
ciertos fármacos por precisar largos espacios de tiempos.
BIBLIOGRAFÍA
- Paul LC et al. Diagnostic criteria for chronic
rejection/accelerated grafo atherosclerosis in heart and kidney
transplants: joint proposal from the Fourth Alexis Carrel. Conference on
Chronic Rejection and Accelerated Arteriosclerosis in Transplanted Organ.
Transplant Proc. 1993;25:2022-3
- Ponticelli C. Progression of renal damage in chronic
rejection. Kidney Int Suppl 2000;75:S62-70
- Rush DN, Karpinski ME, Nickerson P, Dancea S, Birk P,
Jeffery JR. Does subclinical rejection contribute to chronic rejection in
renal transplant patients?. Clin Transplant 1999;13:441-6
- Tejani A, Emmett L. Acute and chronic rejection. Semin
Nephrol 2001;21:498-507
- Stoica SC, Goddard M, Large SR The endothelium in
clinical cardiac transplantation. Ann Thorac Surg 2002;73:1002-8
- Nadasdy T, Smith J, Laszik Z, Waner JL, Johnson LD,
Silva FG. Absence of association between cytomegalovirus infection and
obliterative transplant arteriopathy in renal allograft rejection. Mod
Pathol. 1994;7:289-94
- Womer KL, Lee RS, Madsen JC, Sayegh MH. Tolerance and
chronic rejection. Philos Trans R Soc Lond B Biol Sci 2001;356:727-38
- Paul LC Glomerular hypertension an under-appreciated
aspect of chronic rejection. Nephrol Dial Transplant 2001;16:213-5
- Kauppinen H et al. Sequential analysis of adhesion
molecules and their ligands in rat renalallografts during the development
of chronic rejection. Transpl Int 2000;13:247-54
- Labarrere CA, Pitts D, Nelson DR, Faulk WP. Coronary
artery disease in cardiac allografts: association with arteriolar
endothelial HLA-DR and ICAM-1 antigens. Transplant Proc. 1995
Jun;27(3):1939-40
- Lautenschlager I et al. Human herpesvirus-6 infection
is associated with adhesion molecule induction and lymphocyte infiltration
in liver allografts. J Hepatol 2002;37:648-54
- Viklicky O, Kvasnicka J, Teplan V, Vitko S.
Atherogenesis markers in patients with chronic rejection of renal
allografts. Ann Transplant 2001;6:16-8
- Laine J, Etelamaki P, Holmberg C, Dunkel L. Apoptotic
cell death in human chronic renal allograft rejection. Transplantation
1997;63:101-5
- Nieuwenhuis P, Hillebrands JL, Rozing J. Chronic
allograft rejection associated vasculopathy and synthetic biodegradable
vascular grafts: a lesson to learn?. Crit Rev Immunol 2000;20:85-8
- Labarrere CA, Nelson DR, Park JW. Pathologic markers
of allograft arteriopathy: insight into the pathophysiology of cardiac
allograft chronic rejection. Curr Opin Cardiol 2001;16:110-7
- Valantine-von Kaeppler HA. Prevention of chronic
heart allograft rejection. Transplant Proc. 1999;31:1288-9.
- Orloff MS, et al. Prevention of chronic rejection and
graft arteriosclerosis by tolerante induction. Transplantation.
1995;59:282-8.
- Arnold AN, Anaise D, Miller F, Waltzer WC, Rapaport
FT. Prevention of chronic rejection by cyclosporine and prednisone.
Transplant Proc. 1987;19:2122-3
- Yusuf-Makagiansar H, Anderson ME, Yakovleva TV,
Murray JS, Siahaan TJ Inhibition of LFA-1/ICAM-1 and VLA-4/VCAM-1 as a
therapeutic approach to inflammation and autoimmune diseases. Med Res Rev
2002;22:146-67
- Nowaczyk M, Gorski A, Durlik M, Wyzgal J, Perkowska-Francka
A Role of adhesion molecules in chronic allograft rejection. Arch Immunol
Ther Exp (Warsz) 1999;4):373-5
[Programa
del Congreso] [Índice del Curso]
[Cursos Cortos] [Seminarios]
|
. |