J. FARIŃA GONZALEZ Y MC. MILLANA DE YNES
![]()
| [Índice] [PCR] [Aplicaciones de la PCR] [Bibliografía] |
Las aplicaciones de la PCR son múltiples, desde la arqueología, la Medicina forense y la Medicina clínica. Permite el diagnóstico de enfermedades infecciosas en unas horas frente a la espera de los cultivos. Además, las cantidades necesarias son mínimas, por lo que ese inconveniente es obviado. También tiene esta técnica interés para la demostración de genes indicadores de diversos rasgos que conduzcan a patología. Del máximo interés parece ser la demostración de oncogenes, genes supresores de tumor que en oncología van teniendo valor en el estudio pronóstico de ciertos tumores. Estudios de Biología molecular han demostrado que las células tumorales del carcinoma de pulmón han adquirido lesiones genéticas como activación de oncogenes dominantes o la inactivación de oncogenes supresivos. En relación con los oncogenes dominantes estas lesiones se manifiestan como mutaciones puntuales en las regiones que codifican los oncogenes de la familia ras, principalmente el gen K-ras en el adenocarcinoma de pulmón. Las mutaciones de los genes de la familia ras condicionan el cambio de un solo aminoácido en la proteína que estos genes codifican, generalmente en las posiciones 12, 13, 59, 61 ó 53. Estas alteraciones se descubren hasta en el 30% de los adenocarcinomas de pulmón mediante oligonucleótidos sintéticos tras la amplificación del ADN por la reacción en cadena de la polimerasa.
También pueden aparecer amplificación, redistribución o pérdida de control de la transcripción de los oncogenes de la familia myc, con mayor frecuencia el c-myc en los carcinomas de células pequeńas. Probablemente en los próximos ańos asistiremos a nuevas aplicaciones pronósticas y diagnósticas mediante el desarrollo de estas técnicas.
A continuación exponemos los pasos que seguimos en el Laboratorio de Patología Molecular del Hospital Clínico: La amplificación del ADN mediante la técnica de PCR se realiza toda ella en un sólo tubo de eppendorf de 0,5 ml., que contendrá: ADN problema, oligonucleótido cebadores o primers, desoxinucleótidos trifosfatos libres, Taq polimerasa, magnesio y tampón de reacción.
Hoy día esta técnica se ha simplificado y automatizado gracias a la aparición del termociclador, y diferentes Kits que han surgido en el mercado. El primer paso realizado en el termociclador es la desnaturalización del ADN, para ello se eleva la temperatura a 94şC conseguiéndoos así la separación de las dos hebras del ADN. Hay que tener la precaución de colocar los tubos de amplificación después de que el aparato haya alcanzado dicha temperatura, para evitar uniones inespecíficas que provocarían la amplificación de fragmentos de ADN no diana, y habrá que mantener un tiempo de desnaturalización suficiente, ya que aunque ésta es muy rápida (segundos) la temperatura tarda un tiempo en alcanzarse en el interior del tubo y esto nos conducirá a desnaturalizaciones incompletas con el consiguiente fallo de amplificación. Si, por el contrario, este tiempo se alarga demasiado puede disminuir la actividad de la Taq polimerasa reduciéndose la eficacia de la amplificación.
Para amplificar un fragmento específico de ADN molde necesitamos dos olgonucleótidos cebadores distintos, no complementarios que se van a unir a los extremos de cada cadena de ADN diana que queremos amplificar, una vez desnaturalizado. Los oligonucleótidos cebadores se sintetizan químicamente de forma automática, pero para ello se debe conocer previamente la secuencia de ADN que se quiere amplificar o por lo menos la región a ambos extremos del fragmento ADN diana. Estos cebadores presentan una longitud aproximada de 20 nucleótidos , ya que cebadores más pequeńos aumentan el número de uniones inespecíficas dando lugar a la amplificación de segmentos del ADN no diana y oligonucleótidos mayores son difíciles de sintetizar.
Una vez unidos el ADN diana y el cebador, la Taq polimerasa se encarga de ir ańadiendo los desoxinucleótidos trifosfatos libres al extremo hidroxilo 3´ del oligonucleótidos cebador para formar la cadena complementaria del ADN, esta elongación siempre en sentido 5´ ® 3´, es muy rápida, pues la enzima es capaz de incorporar unos 150 nucleótidos por segundo. La actividad de la enzima depende de la concentración de magnesio en el tampón de la reacción, ya que éste influye en la unión el cebador con el ADN molde y también en la disociación de las cadenas de ADN; un exceso de magnesio conducirá a un acumulo de productos no específicos, mientras que su defecto disminuye de forma importante la cantidad de secuencias sintetizadas.
En citopatología respiratoria la aplicación más frecuente es la demostración de gérmenes que afectan a pacientes inmunodeprimidos que precisan de una terapéutica rápida. Las muestras pueden ser recogidas de torundas de algodón, desde líquido pleural, cepillado bronquial, esputo, BAL, PAAF o de material incluido en parafina.
La amplificación de fragmentos específicos del ADN mediante PCR produce de forma rápida y eficaz muchas copias de ADN que permiten un diagnóstico rápido de tuberculosis, papilomavirus, citomegalovirus y clamidias entre otros gérmenes.
Las torundas utilizadas deben ser estériles sin ningún medio para la fijación ni transporte. Las células se obtendrán agitando en un vórtex con suero salino durante un minuto. Inmediatamente se decanta el sobrenadante a un tubo de eppendorf de 1,5 m autoclavado y se centrifuga a 12.000 rpm durante diez minutos. Se retira el sobrenadante y se resuspende el precipitado de células en 100 ml de una mezcla que contiene Tween 20, proteinasa K y agua destilada que se incuba en un bańo a 55ş C durante tres horas. Transcurrido este tiempo es necesario inactivar la proteinasa k hirviendo las muestras a 100ş C durante diez minutos. Se centrifuga a 12.00 rpm otros diez minutos y el sobrenadante que contiene el ADN se cambia a otro tubo de eppendorf autoclavado del que se tomará una alicuota de 5 ml para proceder a la reacción de amplificación. Esto último es común para todos los tipos de muestras que se quieran amplificar. El resto del sobrenadante, que contiene el ADN, se podrá guardar a 4ş C o a -20ş c para posteriores estudios si fuese necesario.
Si se inhibe la reacción de amplificación se debe proceder a la purificación de la muestra partiendo del ADN que se había guardado, mediante resinas de purificación que concentran la muestra y eliminan inhibidores de la Taq polimerasa. Para ello, se ańade al ADN extraído (100-300 ml) de la muestra 1 ml de resina de purificación, se mezcla el contenido del tubo invirtiendo el tubo varias veces y se deja incubar durante al menos 5 minutos a temperatura ambiente.
Después se hace el lavado de la resina, para lo que necesitamos unir las columnas de purificación previamente marcadas con el número de muestra, a jeringuillas de 2 ml a las que se ańade el contenido de cada tubo de muestra. El líquido se pasa lentamente (1 gota/segundo) por la columna utilizando el émbolo de la jeringuilla. Una vez eluida la columna, se separa de la jeringuilla, para evitar arrastrar la resina al sacar el émbolo y se vuelve a fijar la columna al extremo de la jeringuilla. A continuación se lava la resina con 2 ml de solución de lavado, aplicando presión sobre el émbolo de la jeringuilla y cuando se ha pasado toda la solución de lavado se coloca la columna en un tubo de eppendorf y se centrifuga 20 segundos a 12.000 rpm para eliminar los restos de la solución de lavado.
Por último se hace la elución del ADN, para ello la columna seca se pasa a otro tubo de eppendorf, previamente marcado con el número de muestra, y se ańaden 50 ml de solución de elución a la columna, se deja incubar durante al menos 5 minutos para que el ADN se despegue de la resina. Finalmente, se eluye el ADN por centrifugación a 12.000 rpm durante 20 segundos. De este ADN extraído, unos 50 ml, se toman 5ml para la reacción de PCR y el resto se puede guardar a 4şC ó -20şC para posteriores estudios.
Si se parte de un cepillado bronquial las muestran han de centrifugarse durante 10 minutos a 3.000 rpm y retirar el sobrenadante, el precipitado se resuspende en agua destilada y se pasa a un tubo limpio de eppendorf. Después se vuelve a centrifugar 5 minutos, aunque esta vez a 12.000 rpm y se retira el sobrenadante al que se le ańaden 100 ml de proteinasa k para seguir los mismos pasos que en el párrafo anterior aunque en este caso no suele ser necesario purificar la muestra.
Cuando las muestras son esputos o BAL deben ser clarificadas y descontaminadas antes de la extracción del ADN. En nuestro laboratorio la clarificación del esputo se realiza incubado a 37ş C con lauril sulfato de sosa, se centrifuga a 3.000 rpm y se decanta; el precipitado se neutraliza ańadiendo ácido ortofosfórico con bromocresol púrpura. A continuación para inactivar microorganismos patógenos se incuban las muestras a 75şC durante una hora y se toman 0,5 ml de muestra para la extracción del ADN. Estos 0,5 ml se ponen en un tubo de eppendorf y se centrifugan durante 5 minutos a 12.000 rpm, se decanta el sobrenadante y el precipitado se lava con 1 ml de agua destilada. El precipitado se recentrifuga y resuspende en 100 ml de proteinasa k, de incuba 1 hora a 56şC y después hierven durante 10 minutos, se centrifuga otros 5 minutos a 12.000 rpm y se separa el sobrenadante a un tubo de eppendorf. El ADN extraído debe ser purificado como en el párrafo anterior, antes de la reacción de amplificación.
Cuando la muestra es un líquido pleural partimos de 0,5 ml y se trata igual que el cepillado, con la salvedad de que éste previamente se debe congelar y descongelar durante 2 minutos a 37şC dos veces.
Si partimos de un bloque de parafina se deben hacer entre uno y tres cortes de 5 micras, dependiendo del tamańo de la pieza, y se depositan en un tubo de eppendorf donde se incuban 3 horas con proteinasa k y Tween 20 a 55ş C. Llegado a este punto se continúa de igual forma que en los casos anteriores.
El análisis y tipado del ADN una vez amplificado suele realizarse mediante electroforesis, para ello se disponen de distintos medios aunque los más usados son los geles de agarosa y poliacrilamida; dependiendo del número de pares de bases que contengan los fragmentos de ADN que se quieren estudiar se escogerá uno u otro tipo.
La agarosa es un polisacárido purificado del agar que usado a concentraciones del 3% al 1,5% separa fragmentos de entre 200 y 2.000 pares de bases; aunque existen agarosas de mayor resolución como la Metaphor para fragmentos más pequeńos (50 a 500 pb). Para hacer un gel de agarosa se debe disolver en un matraz una solución electrolítica, TBE 1X (Tris-borato-EDTA), con bromuro de etidio y se calienta (mejor en microondas a potencia media) agitando cada 15 segundos, hasta que quede transparente y sin burbujas de aire. Después se vierte en la bandeja de electroforesis cerrada con cinta adhesiva, se coloca el peine para hacer los pocillos, se deja que gelifique durante quince minutos y se pasa a 4şC durante treinta minutos. Una vez gelificado se retira el peine y se pone en la cubeta de electroforesis que contiene como tampón TBE 1X con bromuro de etidio. A continuación se cargan las muestras amplificadas a las que ańadimos azul de bromofenol (colorante de peso molecular conocido que indica la progresión del gel) en los pocillos reservando el primero y el último para un marcador de peso molecular conocido que nos servirá de referencia para estudiar las muestras problemas, y otros dos para un control negativo y positivo que avisara de posibles contaminaciones y del funcionamiento de la reacción de amplificación respectivamente. Es entonces cuando se conecta la fuente de electroforesis, el voltaje aplicado al gel dependerá del tamańo del mismo, unos 80 Voltios para una cubeta de 50 ml.
Cuando se aplica una diferencia de voltaje entre los dos extremos de un gel, los fragmentos de ADN que poseen una carga negativa debido a los restos fosfatos, migran hacia el polo positivo y por lo tanto los pocillos, lugares de carga de los productos amplificados obtenidos al retirar el peine, deben de estar situados en el polo negativo. La intensidad de la corriente determinará la separación de los fragmentos de ADN y éstos se irán depositando en el gel según su peso molecular.
Las enzimas de restricción fueron descritas en microorganismos y han permitido
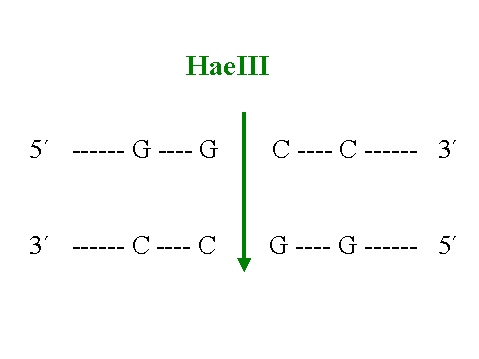
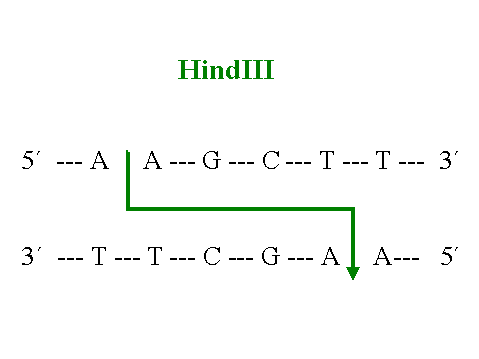
el estudio y manipulación de la patología molecular. Estas enzimas pueden ser endonucleasas o exonucleasas dependiendo de su lugar de acción. Las endonucleasas de restricción son capaces de reconocer una secuencia específica de oligonucleótidos en el interior del ADN bicatenario y excindir la molécula en dicho punto. Las exonucleasas, por el contrario son menos especificas y cortan los extremos del ADN sin necesidad de reconocer una determinada secuencia de bases. Dependiendo del número de pares de bases reconocido por cada enzima los fragmentos de ADN resultantes serán de diferent es tamańo; así endonucleasas que reconocen cuatro pares de bases (figura 4) dividen el ADN en fragmentos menores que aquellas que reconocen secuencias de seis pares (figura 5).
Figura 4. Zona de restricción para el enzima HaeIII.
Figura 5. Zona de restricción para el enzima HindIII.
Gracias al empleo de una o múltiples enzimas de restricción se pueden realizar estudios de segmentos del ADN y definir un mapa detallado de puntos.
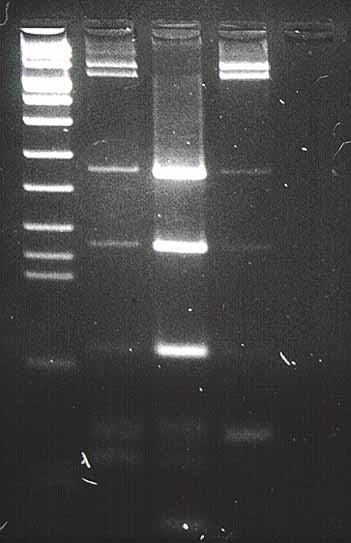
Cuando el frente de azul de bromofenol, ańadido a las muestras, alcanza el final del gel, se podrá apagar la fuente de electroforesis y colocar el gel bajo la ultravioleta de 260 nm ya que el bromuro de etidio tińe los ácidos nucléicos al intercalarse entre sus bases y hace que los fragmentos de ADN a estudio se hagan fluorescentes cuando se exponen a luz ultravioleta, lo que permitirá identificar una o más bandas de diferentes pesos moleculares. Para un control y estudio posterior se hará una foto del gel sobre la luz ultravioleta (
figura 6).
Figura 6. Fotografía del gel de agarosa, sobre luz ultravioleta, usando enzimas de restricción sobre el producto amplificado.
Los geles de poliacrilamida se usan para separar fragmentos de ADN de pequeńo tamańo, pues tiene mayor resolución pudiendo separar bandas con tan solo una base. Cuando ya están mezcladas las moléculas de acrilamida se vierten entre dos placas de vidrio selladas por tres de sus cuatro lados. A diferencia de los geles de agarosa que se hacen con varios milímetros de espesor, estos tienen menos de un milímetro.
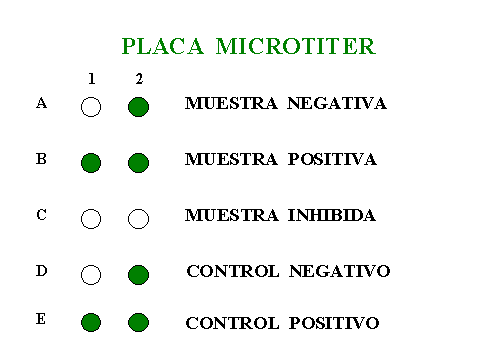
Otra forma de detectar los productos amplificados que con frecuencia se utiliza en nuestro laboratorio es la hibridación con sondas no radiactivas como la digoxigenina o la biotina. Para la detección el producto amplificado marcado con moléculas de digoxigenina, se usa el formato de placa microtiter mediante hibridación con un oligo inmovilizado en el pocillo. Si la secuencia de este oligo es complementaria a la del producto amplificado aparecerá un cambio de color que permitirá su identificación.La valoración colorimétrica del producto amplificado unido al pocillo se realizará utilizando un anticuerpo conjugado con peroxidasa y se medirá en un lector de placas de ELISA. Este método de detección a diferencia de los geles no valora el tamańo del producto amplificado, aunque si puede cuantificar la cantidad del mismo (figura 7).
Figura 7. Esquema que sintetiza los resultados
de la amplificación en una placa microtiter.
Los laboratorios de patología molecular deben tener como mínimo dos zonas o laboratorios independientes, una de extracción del ADN y otra de detección y tipado para evitar la contaminación de las reacciones con cadenas molde de otros orígenes, debido a la alta sensibilidad de la PCR y la abundancia de ADN después de las amplificaciones. Si es posible también debe tenerse una tercer zona para la amplificación de las muestras, donde colocaremos el termociclador. Debido a estos problemas de contaminación además de estas condiciones físicas hay que:
1. Utilizar pipetas con puntas desechables con filtro y deplazamiento positivo, diferentes antes y después de la amplificación.
2. Descontaminar superficies con una solución acuosa de hipoclorito sódico.
3. Preparar la mezcla de reactivo común para varias muestras y posterior distribución a cada una de ellas.
4. Medidas asépticas con flujo laminar y luz ultravioleta en la zona de extracción del ADN.
5. Realizar la amplificación con el mínimo número de ciclos que permitan obtener un resultado óptimo.
6. Abrir con precaución los tubos ya amplificados para evitar la formación de aerosoles.
7. Uso de guantes que deben cambiarse con frecuencia y obligatoriamente cuando se pasa de un área a otra.
8. Es aconsejable también la utilización de gafas y gorros porque los operadores son portadores de productos amplificados.
9. Hay que tomar las precauciones adecuadas cuando se manejan sustancias como el bromuro de etidio que es mutagénico y la poliacrilamida que es neurotóxica.
10. Una vez que la contaminación se ha detectado deberá pararse el trabajo hasta que se efectúe una limpieza, para ello es necesario eliminar los reactivos sospechosos, reemplazar el material contaminado, e incluso puede llegar a ser necesario, la utilización de otras parejas de cebadores para amplificar otras zonas del ADN diana.
| [Índice] [PCR] [Aplicaciones de la PCR] [Bibliografía] |