
 Nº 805. Comunicación libre
Nº 805. Comunicación libre
|
ANTONIO URBAN RAMON[1], OLGA GARCIA VIDAL[1], CONCEPCION MURCIA MESA[1] |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
lay:none">fiogf49gjkf0dCaso clínico inusual de tumor mesenquimal maligno primario de piel del canal auditivo constituido por una proliferación de células monótonas, pequeñas y redondas con inmunoexpresión para CD99, en una paciente de 16 años de edad que acudió a nuestro centro hospitalario por hipoacusia del odio derecho, con el diagnóstico clínico de pólipo inflamatorio. El estudio histológico mostró un tumor de células azules, pequeñas y redondas, con la presencia de células gigantes osteoclásticas-like. Se realiza la discusión de este tipo de tumores y el diagnóstico diferencial, que concluye con la compatibilidad de un sarcoma de Ewing primario de la dermis cutánea del conducto auditivo externo, con la peculiaridad de acompañarse de células de tipo osteoclásticas-like.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Las neoplasias de células azules, pequeñas y redondas son un grupo de tumores heterogéneos que pueden afectar a la región de la cabeza y cuello, dentro de los cuales está el sarcoma de Ewing. Los tumores malignos que propiamente se originan en el conducto auditivo externo son inusuales y la mayoría están relacionados con neoplasias que proceden de las estructuras anexiales de la piel que recubre el canal auditivo. Presentamos un caso excepcional compatible con sarcoma de Ewing extraóseo primario de piel, con el diagnóstico clínico inicial de pólipo inflamatorio y su posterior discusión.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Presentación del caso clínico: Paciente de 16 años de edad de sexo femenino con antecedentes de sinusitis crónica que acudió a nuestro centro hospitalario por hipoacusia del oído derecho de un mes de evolución. El examen del oído evidenció una lesión polipoide de Fueron remitidos fragmentos focalmente de aspecto polipoide, de tejido blando y de coloración blanco-rosada, que agrupados mostraba un tamaño global de 1,4x1 cm. de diámetros máximos. La histología puso de manifiesto un tumor del estroma dérmico con ulceración focal de la epidermis suprayacente. Esta lesión estaba constituida por una proliferación en sábana de células pequeñas y monomorfas, con unos núcleos redondos vesiculados, cromatina fina y algunos con nucleolo inconspicuo, y de citoplasma escaso, pálido y pobremente definido (Fig. 1 y 2), algunos con material de glucógeno (PAS positivo, PAS diastasa negativo). Numerosas figuras de mitosis. Asimismo, en zonas el tumor adquiría un patrón pseudorosetoide (Fig. 3) y otras áreas más edematosos donde se evidenciaban células gigantes osteoclásticas-like entre las células tumorales (Fig. 4 y 5). La lesión se catalogó dentro del grupo de tumores de células azules, pequeñas y redondas. Inmunotinción mostró positividad de las células neoplásicas para vimentina y OK13 (CD19) (Fig. 6), y negatividad para el resto de los marcadores realizados: epiteliales (EMA, CEA, CAM5.2), linfoides y neuroendocrinos (cromogranina y sinaptofisina), neurofilamentos, desmina, enolasa, HMB-45, bcl-2, CD34 y para la proteína S-100. El CD68 mostró únicamente inmunoexpresión de las células gigantes osteoclásticas-like (Fig. 7). El índice de proliferación tumoral Ki-67 fue superior del 50%.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Las neoplasias que se originan del oído externo suelen ser lesiones neoplásicas benignas y muy ocasionalmente malignas, la mayoría derivadas de la piel que recubre conducto auditivo externo. Los sarcomas que afectan al conducto auditivo externo normalmente proceden de estructuras adyacentes o del odio medio e interno. Los sarcomas representan el 15% de todas las neoplasias del organismo y menos del 1% de los sarcomas se localizan en la región de la cabeza y cuello, siendo el más frecuente el rabdomiosarcoma que suele presentarse en las dos primeras década de la vida, con mucha menor incidencia otros sarcomas, como el sarcoma de Ewing/tumor neuroectodérmico primitivo periférico (ES/PNET) (1,2). El sarcoma de Ewing/tumor neuroectodérmico primitivo periférico es un tumor raro que afecta a la población infantil y juvenil con un pico de incidencia en la segunda década de la vida. Pocos casos se han descrito que sean metastáticos a la piel o primarios cutáneos de origen en dermis superficial (3). El caso que nos ocupa exhibe características histológicas y de inmunotinción, CD99 positivas, propias a las células pequeñas y redondas del sarcoma de Ewing. El sarcoma de Ewing y el tumor neuroectodérmico primitivo periférico son dos espectros de una misma entidad tumoral, basado en la clínica, inmunohistoquímica y perfiles citogenéticas. El sarcoma de Ewing afecta más a los huesos y es más indiferenciado con respecto al PNET. Este último, frecuentemente afecta a los tejidos blandos y exhibe más pronunciadas características neuroendocrinas(4). El sarcoma de Ewing extraóseo se describió por primera vez en 1975, la mayoría de ellos en las partes blandas de la región paraespinal, pared torácica y extremidades inferiores, raramente en el tejido subcutáneo. Los sarcomas de ES/PNET como el rabdomiosarcoma pertenecen al grupo de tumores de células azules, pequeñas y redondas caracterizadas por una proliferación de células indiferenciadas relativamente pequeñas y monótonas, de núcleo redondo y vesiculados, con escaso citoplasma (5). Por consiguiente, el diagnóstico diferencial se planteó básicamente con este grupo de lesiones, y en especial con el rabdomiosarcoma alveolar, el linfoma no Hodgkin, el melanoma maligno, el tumor neuroendocrino cutáneo (tumor de células de Merkel) y el osteosarcoma de célula pequeña. El panel inmunohistoquímico (tabla 1.) junto a algunos datos histológicos, como es la presencia de material osteoide en el osteosarcoma, ayuda a descartar las diferentes entidades referidas, y orienta el diagnóstico histológico hacia el ES/PNET primario o metastático. Del mismo modo, la ausencia de expresión neuroendocrina y la positividad en zonas de un patrón membranoso del OK13 (CD99), con la ausencia de otras lesiones alejadas o adyacentes al oído, determina la compatibilidad con el diagnóstico de sarcoma de Ewing extraóseo primario del canal auditivo (3). El OK13 (CD99) es una glucoproteína producida por el gen MIC2 que fue originalmente utilizado como marcador específico del ES/PNET, pero en la actualidad ya se ha argumentado en otro tipo de neoplasias, ciertos tumores neuroendocrinos, linfomas linfoblásticos y en algunos rabdomiosarcomas y melanomas (6). No obstante, la combinación con el resto de los marcadores expuestos en la tabla 1 hacen posible diferenciar estas neoplasias con relación al sarcoma de Ewing. Otro tumor, considerado benigno, que expresa OK13 (CD99) es el tumor fibroso solitario extrapleural. Un caso recientemente se ha publicado en el canal auditivo, con una clínica de lesión polipoide semejante al caso que describimos, aunque con patrón histológico de predominio fusocelular, junto a un estroma focalmente mixoide, y sin actividad mitótica (7). Sin embargo, algunos tumores fibrosos solitarios manifiestan un patrón epitelioide (8) y otros autores han descrito zonas de transformación maligna, con una mayor densidad celular, atipia, focos de necrosis y marcada actividad mitótica, características histológicas que pueden aparentar un tumor de células pequeñas y redondas (9). A diferencia del sarcoma de Ewing las células proliferativas del tumor fibroso solitario expresan además inmunotinción para CD34 y bcl-2. El presente tumor localizado en el canal auditivo de células azules, pequeñas y redondas CD99 positivas, muestra un patrón de crecimiento difuso hipercelular con zonas de patrón pseudorosetoide, afectando únicamente al estroma dérmico y respetando la epidermis suprayacente. Histología e inmunotinción semejante a los pocos casos descritos de sarcomas de Ewing cutáneos primarios de piel, con todo se diferencia del resto por la presencia de las células gigantes osteoclásticas-like. Las células gigantes osteoclásticas-like están entremezclas con las células neoplásicas, constituyendo un carácter reactivo asociado a la lesión tumoral, al igual que sucede con otras lesiones mesenquimales maligna. Estas células gigantes expresan positividad para el marcador monocítico-histiocitario CD68 y negatividad para marcadores epiteliales y mesenquimales, por lo tanto representan verdaderas células osteoclásticas derivadas probablemente de células precursoras macrofágicas de la médula ósea circulantes en la sangre periférica (10,11). Tabla 1. Panel inmunohistoquímico.
ES: Sarcoma de Ewing ; RABD: Rabdomiosarcoma; OSC: Osteosarcoma; ALC: antígeno leucocitario común.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
En resumen, describimos un tumor mesenquimal maligno de células pequeñas y redondas compatible con sarcoma de Swing primario de piel del conducto auditivo externo, con la peculiaridad de mostrar células osteoclástica-like reactivas, más propia de otros sarcomas. Al igual que sucede en los sarcomas de Ewing primarios de piel descritos hasta la fecha, ostenta una serie de características comunes: su clínica indolente, su presentación en edad juvenil (16 años), la mayoría de los casos descritos con edades inferiores a los 20 años, y un pronóstico excelente, a diferencia de los que ocurre en otras localizaciones, óseas o extraóseas. En la actualidad la paciente lleva nueve años de seguimiento, sin mostrar recidivas ni progresión de la enfermedad neoplásica.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
1. Wanebo H, Koness J, MacFarlane JK, et al. Head and Neck Sarcoma: Report of the Head and Neck Sarcoma Registry. Head & Neck. 1992; 14:1-7. 2. Harman M, Kiroglu F, Kösem M, Ünal Ö. Primary Ewings sarcoma of the paranasal sinus with intracranial extension: imaging features. Dentomaxilofacial Radiology. 2003; 32:343-346. 3. Hasegawa SL, Davison JM, Ruten A, et al. Primary Cutaneous Ewings Sarcoma. Am J Surg Pathol. 1998; 22(3):310-318. 4. Thompson LDR. 5. Meis-Kindblom J, Stenman G, Kindblom LG. Differential Diagnosis of small Round Cell Tumors. Seminars in Diagnostic Pathology. 1996; 13 (3):213-241. 6. Wilkerson AE, 7. Rezk S, Yousef M, Zamansky M, et al. Solitary Fibrous Tumor of the Auditory Canal. Arch Pathol Lab Med. 2004; 128:e169-e171. 8. Warraich I, Dunn DM, Oliver JW. Solitary Fibrous Tumor of the Orbit with Epithelioid Features. Arch Pathol Lab Med 2006; 130:1039-1041. 9. Gidwani AL, Mullan FJ, Kenny B. Solitary fibrous tumour of the falciform ligament containing multiple foci of malignant transformation. J Clin Pathol. 2004; 57:546-547. 10. Mentzel T, Calonje E, Fletcher C. Leiomyosarcoma with Prominent Osteoclast-like Giant Cells. Analysis of Eight Cases Closely Mimicking the So-called Giant Cell Variant of Malignant Fibrous Histiocytoma. Am J Sug Pathol. 1994; 18(3):258-265. 11. Leung KM, Wong S, Chow TC, et al. A Malignant Gastrointestinal Stromal Tumor with Osteoclast-like Giant Cells. Arch Pathol Lab Med. 2002; 126:972-974.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
.jpg) fiogf49gjkf0dCÉLULAS MONOMORFAS, PEQUEÑAS Y REDONDAS, DE NUCLEOS VESICULADOS">
fiogf49gjkf0dCÉLULAS MONOMORFAS, PEQUEÑAS Y REDONDAS, DE NUCLEOS VESICULADOS">
.jpg) fiogf49gjkf0d">
fiogf49gjkf0d">
.jpg) fiogf49gjkf0dIMAGEN PSEUDOROSETOIDE">
fiogf49gjkf0dIMAGEN PSEUDOROSETOIDE">
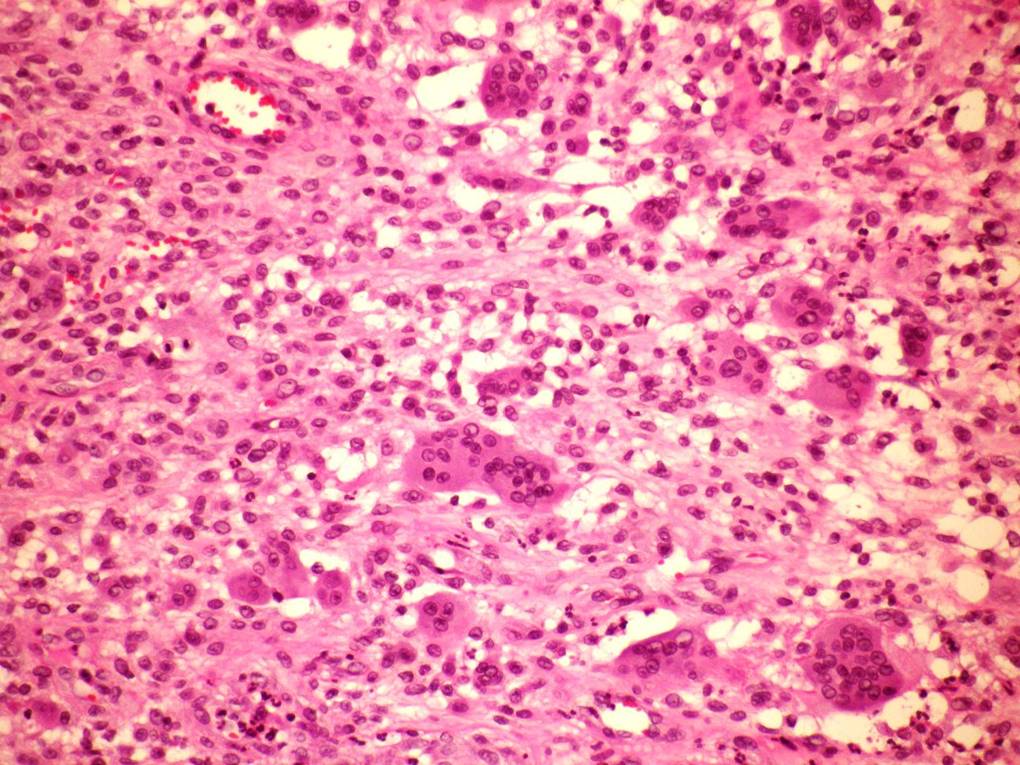 fiogf49gjkf0dCELULAS GIGANTES OSTEOCLÁSTICAS-LIKE">
fiogf49gjkf0dCELULAS GIGANTES OSTEOCLÁSTICAS-LIKE">
.jpg) fiogf49gjkf0dCELULAS GIGANTES OSTEOCLÁSTICAS-LIKE">
fiogf49gjkf0dCELULAS GIGANTES OSTEOCLÁSTICAS-LIKE">
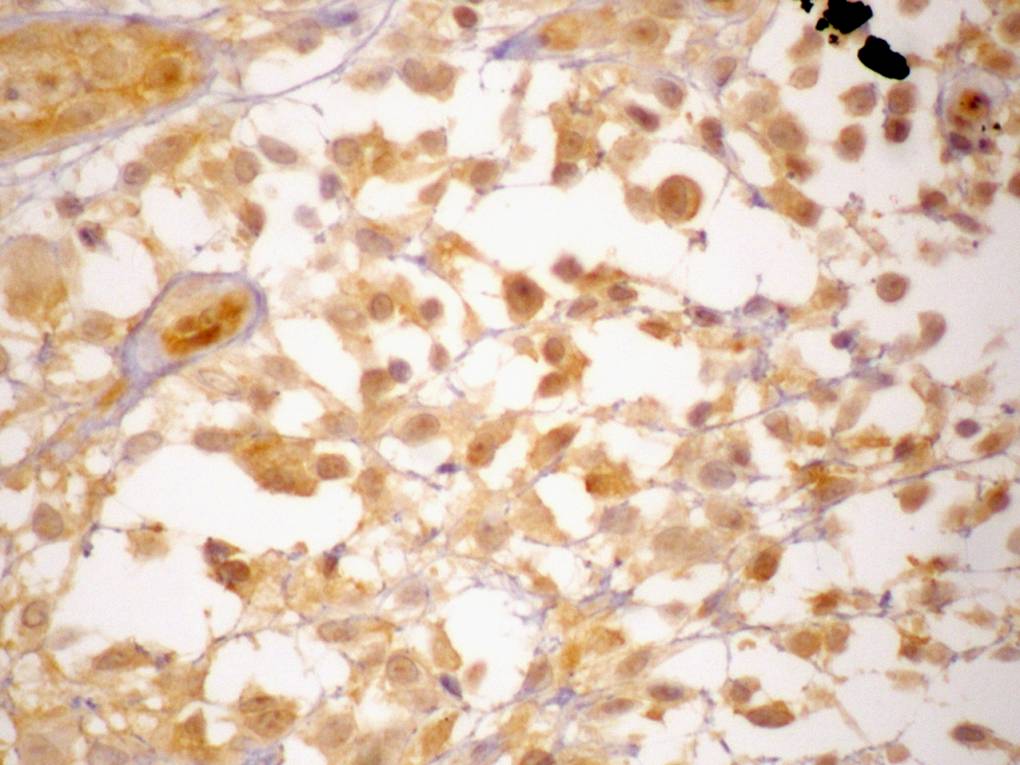 fiogf49gjkf0dOK13 (CD99) POSITIVO EN LAS CÉLULAS TUMORALES">
fiogf49gjkf0dOK13 (CD99) POSITIVO EN LAS CÉLULAS TUMORALES">
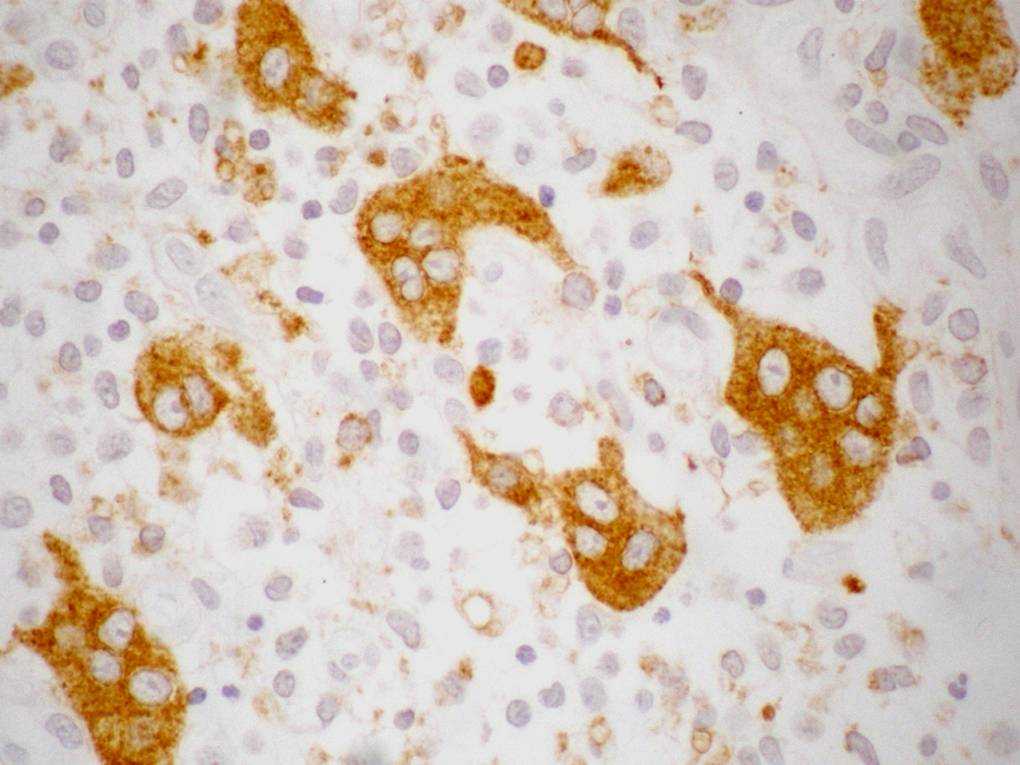 fiogf49gjkf0dCD68 POSITIVO DE LAS CÉLULAS OSTEOCLÁSTICAS-LIKE">
fiogf49gjkf0dCD68 POSITIVO DE LAS CÉLULAS OSTEOCLÁSTICAS-LIKE">
Web mantenido y actualizado por el Servicio de informática uclm. Modificado: 16/06/2015 17:19:20