
 Nº 759. Comunicación libre
Nº 759. Comunicación libre
|
Mirta García Jardon[1], Lech Banach[1], A. Dhaffala[2], Charl Hobson[3], Jeevamoney Govender[1] |
|
|
lay:none">fiogf49gjkf0d Se muestran los resultados de biopsia en un paciente con una masa tumoral de partes blandas del cuero cabelludo, de crecimiento lento, de alrededor de un año de duración, que fué resecada en un varón de 18 años. Clínicamente, el tumor era blando, no doloroso, fijado al cráneo. La TAC de cráneo mostró infiltración del hueso, con penetración en la cavidad craneana. Se realizó biopsia excisional y se envió para estudio a nuestro departamento. Este trabajo presenta los hallazgos clínico-morfológicos (macro y microscópicos) de la biopsia y se realizan algunas consideraciones acerca de su posible histogénesis.
|
||
|
|
Los tumores neuro-ectodérmicos primarios periféricos (TNEPp) (en inglés conocidos como PNETs), son neoplasias de células redondas pequeñas que usualmente se originan ya sea en hueso o partes blandas, incluyendo la pared torácica, cerca de la columna vertebral, pelvis, brazos y piernas. Los mismos pertenecen a la familia del grupo de tumores que incluye al sarcoma de Ewing (1). Son muy raros en la infancia, pero pueden ocurrir en la niñez y más comúnmente en la adolescencia. Los mismos son muy poco frecuentes en descendientes de africanos y asiáticos (1). Los tumores neuro- ectodérmicos primitivos están compuestos de células azules redondas que en la mayoría de los casos muestran translocaciones citogenéticas y una morfología especial inmunohistoquímica (2). Este artículo reporta los hallazgos clínico patológicos en un paciente masculino, africano, de 18 años de edad, con una historia de masa de crecimiento lento, de un año de duración, en el cuero cabelludo.
|
|
|
|
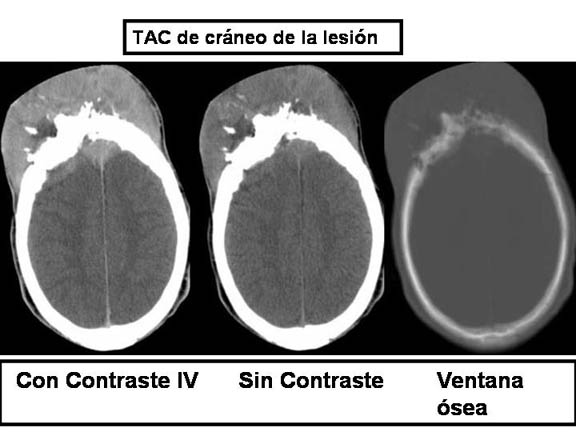
Paciente masculino de 18 años de edad, con historia de una masa de crecimiento lento en el cuero cabelludo, de un año de duración. La TAC de cráneo mostró una masa de tejidos blandos, infiltrando hueso con penetración intracraneal (figura 1) en la región media (biparietal). Se realizó exéresis del tumor, incluyendo partículas de hueso del cráneo, y se envió al servicio de anatomía patológica para su análisis.
Se recibieron dos especímenes quirúrgicos en el servicio, uno pequeño, incluyendo particulas de hueso (no se muestran imágenes del mismo) y el tumor en sí. Un aspecto macroscopico de la masa tumoral se observa en las figuras 2 y 3.
La vista original del reporte de biopsia se ilustra en las figuras 7 y 8, de las cuales se omitieron los datos de identificación personal.
|
|
|
|
Los tumores neuro ectodérmicos primitivos periféricos (en inglés PNETs) son tumores malignos muy raros de células redondas. Los mismos se originan principalmente en piel y/o tejidos blandos y la mayor parte se diagnostica erróneamente con otros nombres (1).
En América, la incidencia de estas neoplasias es de 2.9%/millón de habitantes menores de 20 años. Diez por ciento de ellos aparecen entre 20 y 30 años y casos en edades más avanzadas son infrecuentes.
La supervivencia de los mismos depende grandemente de la presentación inicial y localización o confinamiento del tumor (2). Más de la mitad de estos casos (80%) se presenta con tumor localizado, mientras que en el resto se detectan metástasis en pulmones, hueso y médula ósea. La supervivencia es de un 60%, aunque en casos localizados llega a alcanzar el 70%. Las metástasis confieren un pronóstico muy pobre con una supervivencia de menos del 25%.
Algunos autores consideran enigmática la distribución racial, y en América el tumor es 9 veces más frecuente en blancos que en negros (2), mientras que la neoplasia predomina en el sexo masculino (2). Lo mismo sucede con la edad, aunque se refiere la segunda década como el pico más alto de incidencia (2). Nuestro paciente fue un varón negro de 16 años.
El diagnóstico adecuado se basa en la demostración, por microscopía electrónica de gránulos neuroendocrinos, de una combinación de varias moléculas por biología molecular, y de la positividad a más de un marcador tumoral neural mediante la inmuno-histoquímica. Dichas neoplasias deben diferenciarse de otros tumores malignos cutáneos de células redondas pequeñas. El número de estos casos estudiados completamente es escaso, y aunque no existen conclusiones firmes acerca del comportamiento biológico a largo plazo de los mismos, algunos consideran que una evolución favorable puede ser posible en algunos casos (1).
Nosotros no pudimos hacer citogenética para estudiar el caso, aunque realmente no la consideramos necesaria, ya que encontramos positividad para enolasa neurona-especifica, cromogranina, sinaptofisina y S-100. No en todas las células tumorales encontramos positividad para todos los marcadores utilizados, pero al menos algunas de ellas reaccionaron positivas a los cuatro.
Descrito por primera vez en 1921 como sarcoma, por James Ewing, luego de observar radio sensibilidad en un pequeño subgrupo de tumores óseos. En los inicios de 1980s, se encontraron características comunes entre el sarcoma de Ewing y los tumores neuro-ectodérmicos primitivos primarios; como el hecho de que en ambos existía el mismo tipo de translocación cromosómica t (11; 22) (2). Más tarde se encontraron, durante la misma década, patrones similares de expresión bioquímica y la expresión del mismo oncogen para los dos; con lo cual se empezaron a categorizar como Tumores de la familia del sarcoma de Ewing (2). El grupo incluye el sarcoma de Ewing, el tumor periférico neuroectodérmico primitivo (PNET), el neuroepitelioma, el sarcoma de Ewing atípico y el tumor de Askin ( PNET de la pared torácica) (2). Todos ellos suelen ser manejados similarmente, debido a su semejanza, más que a su variedad histológica (localización o no, metástasis etc.). Nosotros, sin embargo, no hemos recibido retroalimentación ni hemos dado seguimiento al estado de nuestro paciente después de haberlo referido, puesto que una vez diagnosticados, todos se refieren a un hospital oncológico provincial para su manejo.
Aunque el crecimiento de estas neoplasias suele ser rápido, y las mismas se describen como masas blandas y pálidas con necrosis extensa (3), nosotros encontramos una morfología macroscópica similar, pero una historia de un año de crecimiento. En nuestro caso, zonas de necrosis amarillentas y de color arenoso se encontraron alternando con áreas de hemorragia y tejido tumoral viable, blanquecino y homogéneo.
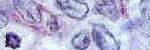
Histológicamente encontramos las células tumorales dispuestas en un patrón trabecular o lobular, con un estroma vascularizado. Las células mostraron citoplasma eosinófilo pálido, escaso, núcleo redondo hipercromático y nucleolo prominente. Estructuras similares a las rosetas de Wright también se observaron (figuras 4 y 5), y la positividad a todos los marcadores neurales mencionados anteriormente. No encontramos sin embargo esa fuerte positividad al PAS como se ha descrito(3), aunque algunas células reaccionaron aisladamente.
Nosotros no tenemos a disposición todos los marcadores tumorales del grupo MIC-2 como para hacer diagnostico diferencial con todos los tumores de células azules (leucemias/linfomas linfoblásticos, rabdomiosarcomas, tumor desmoplástico de células redondas pequeñas, sarcoma sinovial, y otros, que deben ser MIC-2 reactivos (3)), pero tenemos algunos de ellos a disposición (marcadores para músculo, neurales, epiteliales y linfoides), y no encontramos positividad en ninguno de los marcadores no neurales que utilizamos en este caso (EMA, citoqueratina, desmina, actina músculo especifica, CD20, CD 43, CD 45 etc.) (3).
Se ha descrito que la diferenciación neural en este grupo de tumores implica un peor pronóstico independientemente del tipo de tratamiento utilizado (4) y en 2005 se describió un protocolo para el manejo de este tipo de tumores (5). Nosotros no podemos corroborar o negar esto debido a que los casos se remiten a centros especializados.
Sería interesante poder hacer seguimiento en este tipo de pacientes, así como estudiar un número mayor de estos casos.
|
|
|
|
Nuestro agradecimiento al servicio de radiología de nuestro hospital, en especial al la Profesora Targonska, por su ayuda con el manejo de las imágenes radiológicas.
Nuestro eterno agradecimiento al profesor A. Stepien, jefe de nuestro servicio y colega durante 10 años consecutivos, sin cuya valiosa ayuda no hubiesemos tenido suficiente tiempo disponible para realizar este tipo de actividades.
|
|
|
|
1 - Banerjee SS, Agbamu DA, Eyden BP, Harris M. : Clinicopathological characteristics of peripheral primitive neuroectodermal tumour of skin and subcutaneous tissue Histopathology. 1997 Oct;31(4):355-66. 2 - Jeffrey A Toretsky, MD: 3 - 4 - Leonard H. y cols: Neural Differentiation and Prognosis in Peripheral Primitive Neuroectodermal Tumor. Journal of Clinical Oncology, Vol 18, Issue 10 (May), 2000: 2187-2188, © 2000 American Society for Clinical Oncology 5 - Carpentieri D. F. y Cols: Protocol for the Examination of Specimens From Pediatric and Adult Patients With Osseous and Extraosseous
|
|
|
|
- Juan Pablo Garcia de la Torre (11/06/2007 12:46:48)
- Mireyi Meriño Martínez (21/06/2007 14:31:17)
- Mirta Garcia Jardon (21/06/2007 18:01:52)
|
|
|
|
|
.JPG) fiogf49gjkf0dUniversidad Walter Sisulu">
fiogf49gjkf0dUniversidad Walter Sisulu">
.jpg)
.jpg) fiogf49gjkf0dFigura 3: Superficie de corte del tumor. Se observa una apariencia multinodular, algunos hemorrágicos, otros blanquecinos, francamente tumorales y otros amarillentos o color arena de necrosis.">
fiogf49gjkf0dFigura 3: Superficie de corte del tumor. Se observa una apariencia multinodular, algunos hemorrágicos, otros blanquecinos, francamente tumorales y otros amarillentos o color arena de necrosis.">
.JPG) fiogf49gjkf0dFigura 4: Vista panorámica de la neoplasia. Se observa marcada celularidad por pequeñas células azules, de núcleo hipercromático y citoplasma escaso; y la formación de seudorosetas. H/E 10X
">
fiogf49gjkf0dFigura 4: Vista panorámica de la neoplasia. Se observa marcada celularidad por pequeñas células azules, de núcleo hipercromático y citoplasma escaso; y la formación de seudorosetas. H/E 10X
">
.JPG)
.jpg)
.jpg) fiogf49gjkf0dVista superior del reporte original. Datos personales omitidos.">
fiogf49gjkf0dVista superior del reporte original. Datos personales omitidos.">
.jpg) fiogf49gjkf0dFigura 8: Vista inferior del reporte original. Datos personales omitidos.">
fiogf49gjkf0dFigura 8: Vista inferior del reporte original. Datos personales omitidos.">
Web mantenido y actualizado por el Servicio de informática uclm. Modificado: 16/06/2015 17:19:20