

|
MICROSATELLITE INSTABILITY CORRELATES WITH LEVEL OF INFILTRATION IN SPORADIC DESMOID TUMOR Consuelo Junqueira Rodrigues*, Gina Camillo Silvestre*, Aldo Junqueira Rodrigues Junior* |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Background: Desmoid tumor (DT) is a benign soft tissue tumor, characterized by local invasion and frequent recurrences. It arises as a sporadic lesion or as part of familial adenomatous polyposis, which has been associated with mutation in APC gene, responsible for the clearance of b -catenin. Mutant b-catenin is resistant to degradation, and its accumulation could act as an oncoprotein. Microsatellite instability (MSI) may drive tumor progression by promoting the acceleration of mutations in oncogenes and tumor suppressor genes. The present study was undertaken to determine if there is a correlation between clinical & anatomical features of DT and MSI & βcatenin mutation.Material and Methods: Six microsatellite makers (bat 25, bat 26, bat 40, tp53, d2s123 and d17s250) and exon 3 of βcatenin gene were tested in genomic DNA of normal and tumor tissue from 27 patients with sporadic DT.Results: We verified that DT was more common in women and in the second-fourth decades of life. We detected MSI in 85% (23/27) and βcatenin mutation in 68% (15/22) of these patients. We found no correlation between MSI & βcatenin mutation and location of DT. There is a significant correlation between high-frequency of MSI and deeper level of infiltration.Conclusion: The high incidence of MSI and mutant βcatenin in sporadic DT seems to be associated to the benign phenotype, which seems to be dependent on a complex of alternative genetic pathways which may prevent malignancy. However, the aggressive behavior of sporadic DT was associated with high-frequency of MSI.Key words: Desmoid tumor, beta-catenin, microsatellite, MSI
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Desmoid tumor (DT) is a benign soft-tissue tumor, classified among fibromatoses, which is characterized by intense fibroblastic proliferation with minimum microscopic alteration. The infiltrative nature of DT is responsible for its locally aggressive behavior and a high postoperative recurrence rate. The significance of surgical resection with positive resection margins remains controversial as a predictor of local recurrence (Lewis et al 1999). DT can affect individuals of different ages, however it is more common in the second to fourth decades of life, occurring in the abdominal or extra-abdominal wall. It occurs as a sporadic lesion or as part of extraintestinal manifestations of familial adenomatous polyposis, which is associated with germline mutations in the adenomatous polyposis coli (APC) gene (Clark & Phillips 1996). APC protein plays an essential role in the clearance of b -catenin from the cytoplasm. Increased b -catenin may translocate to the nuclei and serve as a transcriptional factor by binding to the T-cell factor/lymphoid enhancer factor family. Mutations in APC or βcatenin genes result in increased cytoplasmic levels of free βcatenin, which may then act as an oncoprotein through constitutive b-catenin-Tcf-regulated transcription (Morin et al 1997).Multiple genetic changes occur during the development of cancer; this stepwise accumulation of abnormalities is likely to be accelerated by genetic instability. The form of genomic instability associated with defective DNA mismatch repair or replication error repair (RER) in tumors is called microsatellite instability (MSI). A panel of five microsatellites has been recommended for research, and tumors may be characterized on the basis of: high-frequency MSI, if two or more of the five markers show instability, and low frequency MSI, if only one marker shows instability (Boland et al 1998). Presence of MSI has been demonstrated in a variety of both hereditary and sporadic tumors. Studies have shown that MSI confers a survival advantage (Lothe et al 1993, Gryfe et al 2000, Maxwell et al 2001), but data is conflicting on its effect on the prognosis of some diseases (MacDonald et al 2000). The present study was undertaken to determine if there is a correlation of clinical and anatomical features with MSI and βcatenin mutation and to identify molecular changes accounting for the phenotypic characteristics of its aggressive behavior.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
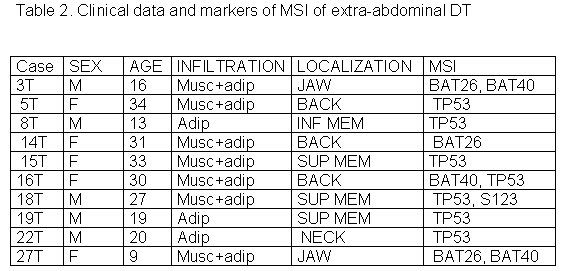
Twenty-seven normal and tumor formalin-fixed, paraffin-embedded tissues were selected from patients of Hospital das Clinicas - School of Medicine - University of São Paulo. Ethics Committee of HC-FMUSP (n 925/01) approved this research protocol. Clinical data of individual tumors are listed in Table 1. Tumor Specimens. Normal and tumor tissues were microdissected using separate 5m m formalin-fixed, paraffin-embedded slides for each one. Tissues were digested overnight at 56o C in a solution containing 10mg/ml proteinase K, and genomic DNA was extracted according to the method previously described (Nascimento, et al. 2003), using Wizard Genomic Kit (Promega).MSI Analysis. Tumor and corresponding normal DNA from each case were analyzed using a panel of six markers. The sequences of the primer pairs were as follows: BAT25 (mononucleotide, 56o C): F= 5-TCG CCT CCA AGA ATG TAA GT-3 and R= 5- GAG CCA TAG TTA AAA TGC AGA-3; BAT26 (mononucleotide, 59o C): F= 5- TGA CTA CTT TTG ACT TCA GCC-3 and R= 5- GGG TTA AAA ATG TTG AAT GGTT-3; BAT40 (mononucleotide, 52o C): F= 5- ATT AAC TTC CTA CAC CAC AAC-3 and R= 5-CAA GGT GGT CTT GCT CTA C-3; D2S123 (dinucleotide, 59o C): F= 5- AAA CAG GAT GCC TGC CTT TA-3 and R= 5- GTC CCA TAG GTG GAA AGT CC-3; D17S250 (dinucleotide, 57o C): F= 5- GGA AGA ATC AAA TAG ACA AT-3 and R= 5- GGT TTA AAT ATA TAT ATG GCC AGC-3; TP53 (dinucleotide, 56oC): F= 5- ACT GCC ACT CCT TGC CCC ATT C-3 and R= 5-CAC CTC GGG CTG AAT AGT ATC CCT-3. PCR reactions were performed under standard conditions in a 25μl volume, 0,2μM primer sense and antisense; +/- 200ng/ml of Genomic DNA; 0,2mM each of dATP, dGTP, dCTP and dTTP; 5U Taq Polymerase Platinum pause 15 minutes (Invitrogem do Brasil); 10X PCR Buffer; 1.5 mM MgCl2. Thermal cycling was performed for 15 minutes (Taq Polymerase Platinum), in cycle 34, 1 minute at 94ºC, 1 minute at annealing, 1 minute at 72ºC and another 10 minutes at 72ºC, stocking at 4ºC. An aliquot of each PCR product was electrophoresed in a 2% agarose gel, stained with ethidium bromide, and observed under ultraviolet light to ensure that a single product of appropriate size was amplified. PCR products were diluted in Stop Solution (95% formamide, 20mM EDTA, 0,05% bromophenol blue and 0,05% xylene cyanol). The diluted solution was boiled at 100ºC and the diluted samples were placed in it for 5 hours, until their cooling, followed by electrophoresis in 8% polyacrylamide gel at 4ºC, 250V, 8W for 1 hour and 30 minutes in the Hoefer 600 SE series in an SDS 1X Buffer. Polyacrylamide gel was reddened in a silver nitrate 0.1% solution and formamide 0.005% for 20 minutes. Detection of MSI determined the presence of new alleles in tumor tissue against normal tissue. B-Catenin gene mutation analysis: was assessed using primer of the exon 3 (F= 5 CTG ATT TGA TGG AGT TGG ACA T-3 and R= 5- CCT TCA GTC AAG AAC AAG TAG C-3). PCR was performed in a 75μl volume containing 0,2μM of sense and antisense primer; +/- 200ng/ml of Genomic DNA; 0,2mM each of dATP, dGTP, dCTP and dTTP; 5U Taq Polymerase Platinum pause 15 minutes; 10X PCR Buffer; 1.5 mM MgCl2. Thermal cycling was performed for 15 minutes (Taq Polymerase Platinum), in cycle 34, 1 minute at 94ºC, 1 minute at 55ºC annealing, 1 minute at 72ºC and another 10 minutes at 72ºC, stocking at 4ºC. Aliquots were purified using the QIAquick PCR purification column (Quiagen, Chatsworth, CA) and sequenced in automated sequencer MEGABASE (Amersham Pharmacia Biotech). Statistical analysis. To elucidate the relationship of MSI and βcatenin with the clinicopathological features of DT, we used Fisher´s Exact Test for univariate analysis. We considered p values below 0.05 statistically significant. Table 1. Clinical data and markers of MSI of abdominal DT
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
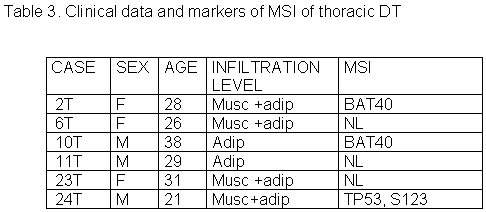
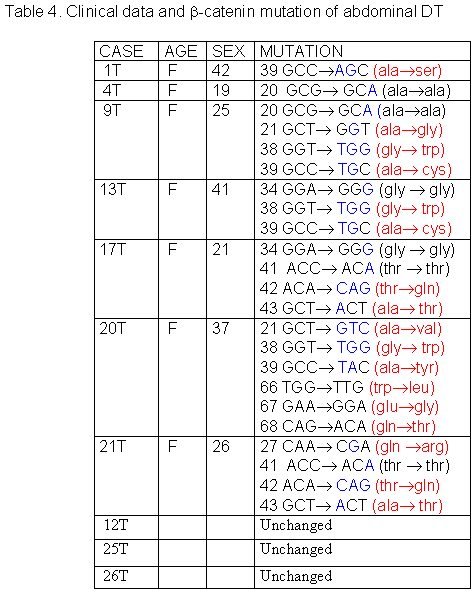
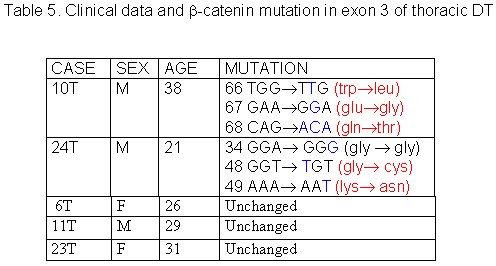
The 27 cases of DT studied were divided in three groups according to tumor site: abdominal, thoracic and extra-abdominal (jaw, back, neck and limbs). The mean age of these patients was 28.2± 9.07 years (min=9 and max=42years), and there was no significant age difference between these three groups of patients. The mean age of DT patients was: abdominal = 32.5± 8.94 years (Table 1), thoracic = 28.8± 5.63 years (Table 2) and extra-abdominal =23.2± years (Table 3). There was no significant age difference between these three groups studied. We demonstrated a significant (p<0.01) association between abdominal DT and women by comparing these three groups of patients. Furthermore, there was a higher prevalence of sporadic DT in women (19/27). MSI. Twenty-seven samples from patients with sporadic DT were tested for the presence of mutations within mono and dinucleotide repeats. The repeats that we studied (BAT25, BAT26, BAT40, D2S123, D17S250 and TP53) were selected to supply optimal information on MSI related to RER defects (Boland et al 1998). We demonstrated that 85% (23/27) of DT presented MSI. Of these DT, 37% (10/27) presented low frequency MSI (MSI-L), and 48% (13/27) presented high frequency MSI (MSI-H). MSI-H was mainly in abdominal and extra-abdominal DT. There was no significant association between location of DT and presence of MSI. There was a significant association (p<0.05) between MSI-H and depth of infiltration when we compared level of infiltration and occurrence of MSI. Therefore, DT with MSI-H showed infiltration at the muscle tissue level. B-CATENIN MUTATION ANALYSIS Fifteen of the 22 (68%) samples of DT analyzed had many point mutations in b -catenin, from codon 20 to codon 68. A great number of DT showed missense mutations in the codon or in neighboring codons of phosphorylation sites, which involved serine/threonine residues: 30 (tyrosine® serine), 34 (glycine® alanine), 38 (glycine® tryptophan), 39 (alanine® cysteine/tyrosine/serine), 42 (threonine® glycine) and 43 (alanine® threonine). The other cases showed missense mutations in sites unrelated to the phosphorylation regions: 21 (alanine® glycine), 26 (glycine® alanine), 27 (glycine® alanine/arginine), 66 (tryptophan® leucine), 67 (glutamic acid® glycine) and 68 (glutamine® threonine). Furthermore, we showed point mutations in codons 20, 34 and 41, where the amino acid was unchanged. We demonstrated that in the same DT there was some kinds of mutations in exon 3 of the b -catenin gene. Six of the ten (60%) cases of abdominal DT showed missense mutations in the neighboring codons of phosphorylation sites (Table 4). Two cases of the five thoracic DT analyzed showed missense mutation, one in the neighboring (codons 48 and 49) and another in unrelated (codons 66, 67 and 68) sites to phosphorylation regions (Table 5). Four cases of the seven extra-abdominal DT showed missense mutations, only two being in neighboring codons of phosphorylation sites (Table 6). We did not find any significant association when we compared the location of DT and depth of infiltration with the presence of b -catenin missense mutation. We verified that the mutation in exon 3 of the b -catenin gene was present in all cases of DT with MSI. Therefore, the cases of DT without MSI presented no b -catenin mutations.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Desmoid tumor occurs with a certain frequency as part of familial adenomatous polyposis (FAP), which is associated with a mutation of the APC gene. The present study involves 27 cases of sporadic desmoid tumors. In these cases, we observed a greater incidence in young adult women, as mentioned in previous accounts (Mungas et al 1976). The pathogenesis of sporadic desmoid tumor not associated with FAP remains unknown, yet a genetic model similar to hereditary neoplasms has been proposed. In the present study, we investigated genetic alterations related to DNA repair caused by MSI, and oncogene activation caused by mutation of β-catenin.To study MSI, we used 06 microsatellite markers, three mononucleotides and three dinucleotides, as suggested by the National Cancer Institute of the USA (Boland et al 1998). We verified that the most sensitive markers were the mononucleotides BAT26 and BAT40, both highly specific for identifying tumors with high-frequency MSI. These findings are in agreement with other studies that verified that mononucleotide microsatellite markers have greater sensitivity and specificity than dinucleotides in detecting defective DNA mismatch repair (Nash et al 2003). We demonstrated that 82% (22/27) of the desmoid tumors presented MSI and therefore they can be associated with defects in the DNA repair pathway, causing an accumulation of mutations in oncogenes and tumor suppressor genes, which would induce early tumor growth (Markowitz et al 1995). This explains the appearance of desmoid tumor in young individuals as seen in our study, where the mean age was 28.2± 9.07. On the other hand, the MSI phenotype also seems to be associated with a good prognosis in endometrial and colorectal cancers. This association may be explained by the loss of DNA repair that favors an alteration in the gene, resulting in a less virulent clinical phenotype (Gryfe et al 2000, Maxwell et al 2001), which can, to some extent, explain the benign character of the desmoid tumor. In the present study, we observed that the abdominal and extra-abdominal (not thoracic) desmoid tumors showed high incidence of MSI, while thoracic tumors did not. Among the cases studied, we demonstrated that 56% presented high MSI, suggesting that these sporadic desmoid tumors can be associated with a familial history of neoplasms. The same was stated by Ruschoff et al (1995): after analyzing MSI studies on patients with sporadic colorectal cancer, they informed that the cases with high MSI were very similar to hereditary colorectal cancer in their morphological and biological characteristics. The association of some cases of desmoid tumors with APC gene mutation and high levels of βcatenin suggest low degradation rate of βcatenin in tumor tissue (Alman et al 1997). Βcatenin is involved in cellular differentiation and proliferation and is quickly degraded in the absence of differentiation or growth signals. APC protein controls degradation through the phosphorylation of serine and threonine residues (Morin et al 1997). Deletion or mutation of one or more βcatenin phosphorylation sites inhibits its degradation, increasing its action as a transcription activation factor. This intracellular increase of βcatenin due to βcatenin or APC gene mutation has been suggested as an important factor in the oncogenesis of colorectal cancer (Gumbiner 1995, Morin et al 1997).Saito et al (2001) verified a correlation between the a ccumulation of βcatenin and the increased expression of the cyclin D1 gene in desmoid tumors, suggesting that βcatenin acted like an oncoprotein in the APC- βcatenin-Tcf pathway, whose target gene is cyclin D1. Yet, this association between βcatenin and cyclin D1 was not correlated to MIB activity, reflecting the benign nature of this tumor. These authors demonstrated a diffuse expression of PCNA in desmoid tumors, suggesting that PCNA is involved in DNA repair and in DNA replication inhibition when it binds to cyclin D1, as stated by Xiong et al (1992). The incidence of βcatenin mutation in benign lesions like desmoid tumor, pilomatricoma and hepatoblastoma suggests that the stabilization of βcatenin is correlated to the benign phenotype. Approximately half of all colon adenomas do not present APC gene mutation but present βcatenin mutation (Kitaeva et al 1997, Sparks et al 1998). B-catenin mutations in cancer have been described and the expression of mutant βcatenin in the cellular nucleus was associated with a non-invasive tumor and favorable prognosis (Mao et al 2001).The wide range mutations in exon 3 of βcatenin detected in the present study affect serine and threonine phosphorylation sites that are important in the degradation of βcatenin and that can lead to an accumulation of βcatenin and translation of oncogenic signals. Additionally, βcatenin mutations occurring at the neighboring sites of Ser/Thr residues, as was observed in the present study, is also resistant to degradation ( Saito et al 2002). Otherwise, these mutations can interfere with the formation of the βcatenin/ά-catenin complex and disrupt the E-cadherin-catenin complex, thereby affecting intercellular adhesion, as well as interfere in the accessibility of serine phosphorylation or in other βcatenin functions, such as when it serves as the substrate for caspase during apoptosis (Aberle et al 1996).In colorectal cancer, a genetic model has been proposed where sequential accumulation of mutations in specific genes, like the APC, K-ras and p53, would transform the normal epithelium in dysplasia and carcinoma. In a study of colorectal cancer, Smith et al (2002) found that only 6.6% of the tumors had a mutation in these three genes while 37.5% had a mutation in only one gene, therefore not confirming that the existence of cancer is a sequential accumulation of mutations. Hence, there seems to be multiple alternative genetic pathways for the appearance of a malignant neoplasia or alternative pathways that prevent a benign neoplasia to become malignant, even if they show genetic alterations related to oncogenes, as is the case with the desmoid tumor. We will show that desmoid tumors affected young adult individuals, were more common in women, and presented a genetic character with a high incidence of MSI and βcatenin mutation. This genetic cancer is very similar to hereditary neoplasms (Liu et al 199 5; Markowitz 2000), which would, in a way, explain its occurrence in young individuals. We will also show a correlation between the kind of infiltration and MSI: tumors located in adipose tissue present low-frequency of MSI while tumors that infiltrate adipose and muscle tissue present high-frequency of MSI.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
CONCLUSION The high incidence of MSI and mutant βcatenin in sporadic DT seems to be associated to the benign phenotype, which is not a result of a sequential accumulation of mutations, but seems to be dependent on a complex of alternative genetic pathways which may prevent malignancy. However, the aggressive behavior of sporadic DT was associated with high-frequency of MSI.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
The present work was sponsored by FAPESP 02/07939-6 and 01/14052-5. We are grateful to Alexandre Silva and Maria das Graças Batista for your technical assistance.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Aberle, H.; Schwartz, H.; Kemler, R. Cadherin-catenin complex: protein interactions and their implications for cadherin function. J. Cell Biochem., 61:514-523, 1996. Alman BA, Pajerski Me, Diaz-Cano S, Wolfe HJ. Increased Beta-catenin protein and somatic APC mutation in sporadic aggressive fibromatoses. Am J Pathol 151: 329-34, 1997. Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, Srivastava. A National Cancer Institute Workshop on microsatellite instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 58:5248-5257, 1998. Clark SK, Phillips RKS. Desmoid in familial adenomatous polyposis. Br J Surg 83:11494-11504, 1996. Gryfe R, Kim IJ, Hsieh ET, Aronson MD, Holowaty EJ, Bull SB. Tumor MIS and clinical outcome in young patients with colorectal cancer N Engl J Med 342: 69-77, 2000. Gumbiner 1995 BM. Signal transduction of βcatenin. Curr Opin Cell Biol 7:634-640,1995. Kitaeva MN, Grogan L, Williams JP, Dimond E, Nakahara K, Hausner P et al Mutations in beta-catenin are uncommon in colorectal cancer occurring in occasional replication error-positive tumors. Cancer Res 57:4478-81, 1997. Lewis JJ, Boland PJ, Leung DHY, Woodruff JM, Brennan MF. The enigma of desmoid tumors. Ann Surg 229:866-873, 1999. Liu B, Farringtion FM, Petersen GM, Hamilton SR, Parsons R, Papadopoulos N, Fukiwata T, Jen J, Kinzler KW and Dunlop MG. Genetic instability occurs in the majority of young patients with colorectal cancer. Nature Med 1: 383-352, 1995. Lothe RA, Peltomaki P, Meling GI et al. Genomic instability in colorectal cancer: relationship of clinicopathological variables and family history. Cancer Res 53:5849-5852, 1993. Mao TL, Chu JS, Jeng YM, Lai PL, Hsu HC. Expression of mutant nuclear b-catenin correlates with non-invasive hepatocellular carcinoma, absence of portal vein spread, and good prognosis. J Pathol 193:95-101, 2001. Markowitz S, Wang J; Myeroff L; Parsons R; Sun L; Lutterbaugh J; Fan RS; Zborowska E; Kinzler KW; Vogelstein B; Brattain M; Willson JKV. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science, 268: 1336-1338, 1995. Markowitz S. DNA repair defects inactivate tumor suppressor genes and induce hereditary and sporadic colon cancer. J. Clin. Oncol., 18:75s-80s, 2000. Maxwell GL, Risinger JI, Alvarez AA, Barret JC, Berchuck A. Favorable survival associated with microsatellite instability in endometrioid endometrial cancers. Obst Gynecol 97: 417-21, 2001. Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of βcatenin in colon cancer by mutations in βcatenin or APC. Science 257:1787-1790, 1997. MacDonald ND, Salvesen HB, Ryan A, Iversen O, Akslen L and Jacobs IJ. Frequency and prognostic impact of microsatellite instability in a large population-based study of endometrial carcinomas. Cancer Res 60: 1750-1752, 2000. Mungas JE, Platz CE, Block GE. Desmoid tumors of the abdominal wall. Surg Clin North Am 56: 207-18, 1976. Nascimento E, Spinelli M, Rodrigues CJ, Bozzini N. Protocolo de extração de DNA de material parafinado para análise de microssatélites em meiomioma uterino. J Bras Patol 39: 253-255, 2003. Nash GM, Gimbel M, Shia J, Culliford AT, Nathanson DR, Ndubuisi M, Yamaguchi Y, Zeng Z, Barany F , Paty P. Automated, multiplex assay for h-frequency microsatellite instability in colorectal cancer. J Clin Oncol 21:3105-3112, 2003. Ruschoff J, Bocker T, Schlegel J, Stumm G, Hofstaedter F. Microsatellite instability: new aspects in the carcinogenesis of colorectal cancer. Virchow Arch 426: 223-27, 1995. Saito T, Oda Y, Tanaka K, Matsuda S, Tamiya S, Iwamoto Y, Tsuneyoshi M. Beta-catenin nuclear expression correlates with cyclin D1 overexpression in sporadic desmoid tumors. J Pathol 195:222-8, 2001. Shields CJ, Winter DC, Kirwan WO, Redmoond HP Desmoid tumors. Eur J Surg Oncol 27:701-6, 2001. Smith G, Carey FA, Beattie J, Wilkie MJ, Lightfoot TJ, Coxhead J, Garner RC, Steele RJ, Wolf CR. Mutations in APC, K-ras and p53 alternative genetic pathways to colorectal cancer. Proc Natl Acad Sci 99:9433-8, 2002. Sparks AB, Morin PJ, Vogelstein B, Kinzler KW. Mutational analysis of the APC/ βcatenin /Tcf pathway in colorectal cancer. Cancer Res 58:1130-4, 1998.Tejpar S, Nollet F, Li C, Wunder Js, Michils G, dal Cin P, Van Cutsen E, Bpat B, van Roy F, Cassiman JJ, Alman BA. Predominance of beta-catenin mutations and βcatenin dysregulation i n sporadic aggressive fibromatoses. Oncogene 18: 6615-20, 1999.Xiong Y, Zhang H, Beach D. D type cyclins associate with multiple protein kinases and DNA replication and repair factor PCNA. Cell 71:505-514, 1992.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
 fiogf49gjkf0dTable 2. Clinical data and markers of MSI of extra-abdominal DT">
fiogf49gjkf0dTable 2. Clinical data and markers of MSI of extra-abdominal DT">
 fiogf49gjkf0dTable 3. Clinical data and markers of MSI of thoracic DT">
fiogf49gjkf0dTable 3. Clinical data and markers of MSI of thoracic DT">
 fiogf49gjkf0dTable 4. Clinical data and -catenin mutation of abdominal DT">
fiogf49gjkf0dTable 4. Clinical data and -catenin mutation of abdominal DT">
 fiogf49gjkf0dTable 5. Clinical data and -catenin mutation in exon 3 of thoracic DT">
fiogf49gjkf0dTable 5. Clinical data and -catenin mutation in exon 3 of thoracic DT">
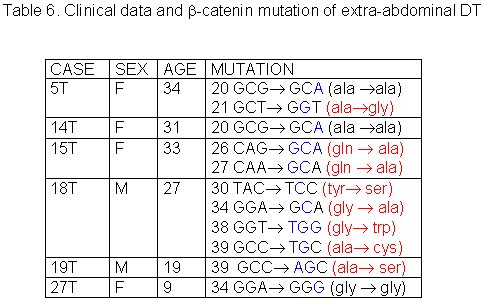 fiogf49gjkf0dTable 6. Clinical data and -catenin mutation of extra-abdominal DT">
fiogf49gjkf0dTable 6. Clinical data and -catenin mutation of extra-abdominal DT">
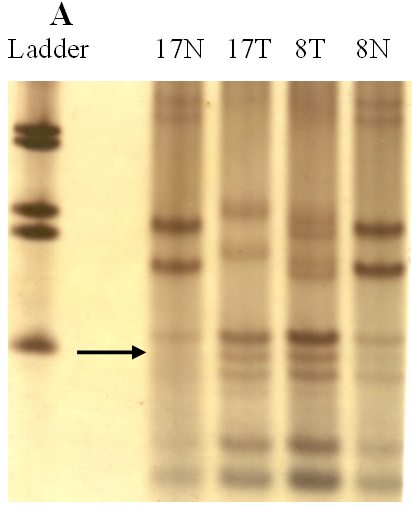 fiogf49gjkf0dFigure 1-A.">
fiogf49gjkf0dFigure 1-A.">
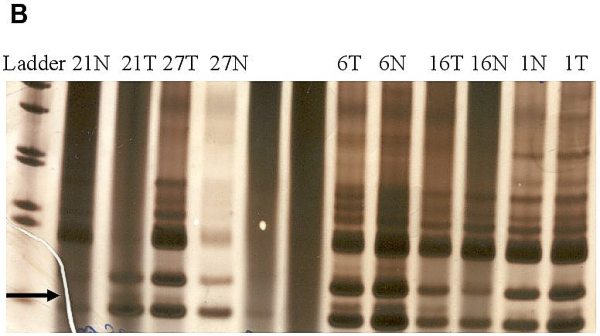 fiogf49gjkf0dFigure 1-B.">
fiogf49gjkf0dFigure 1-B.">
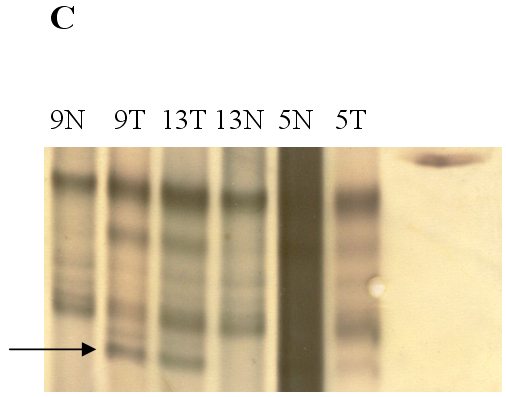 fiogf49gjkf0dFigure 1. Analysis of genetic instability in paired normal (N) and tumor (T) DNA at
loci TP53 -118bp (A), BAT40 123bp (B) and BAT26 132bp (C). In tumor DNAs abnormal patterns indicating deletion are shown at each microsatellite locus. Patients numbers are shown at the lanes.
">
fiogf49gjkf0dFigure 1. Analysis of genetic instability in paired normal (N) and tumor (T) DNA at
loci TP53 -118bp (A), BAT40 123bp (B) and BAT26 132bp (C). In tumor DNAs abnormal patterns indicating deletion are shown at each microsatellite locus. Patients numbers are shown at the lanes.
">
Web mantenido y actualizado por el Servicio de informática uclm. Modificado: 16/06/2015 15:17:25