|
RECIENTES AVANCES EN EL DIAGNÓSTICO Y PRONÓSTICO
DE LOS TUMORES DE CÉLULAS REDONDAS INFANTILES
Samuel Navarro, José Antonio López-Guerrero, Antonio
Pellín, Antonio Llombart-Bosch.
Departamento de Patología, Facultad de Medicina, Universidad
de Valencia, España.
Trabajo realizado gracias a la ayuda FIS 01/0673, Madrid, España.
Los tumores de células redondas constituyen un grupo heterogéneo
de neoplasias que asientan en partes blandas, hueso y órganos
sólidos con una alta incidencia en niños y adolescentes.
Las variedades indiferenciadas de estos tumores plantean problemas
diagnósticos que en muchas ocasiones solo se resuelven aplicando
técnicas de patología molecular o inmunohistoquímica.
Por su frecuencia y por sus características clínico-biológicas
peculiares, merecen destacarse en este grupo el denominado tumor
de Ewing/tumor primitivo neuroectodérmico (Es/PNET), el tumor
desmoplastico de célula pequeña (DSRCT), el neuroblastoma,
el rabdomiosarcoma, el sarcoma sinovial y el condrosarcoma mixoide.
El Es/PNET es una neoplasia heterogénea que representa entre
un 5 y un 15 % de los tumores malignos, que suele afectar a hueso
y partes blandas de niños y adolescentes y cuyo diagnóstico
en las biopsias se basa en la presencia de una celularidad monomórfica,
con elementos de pequeño tamaño, con glucógeno
citoplásmico, y en los casos con abierta diferenciación
neural, por la detección de granulos neurosecretores con
la ultrastructura y la expresión de marcadores neurales (Enolasa,
cromogranina, sinaptofisina, HNK-1, etc) (1).
El diagnostico diferencial con otros tumores de células
redondas se realiza gracias a la detección inmunohistoquimica
de CD99 (HBA-71, MIC2) y a la presencia de un marcador citogenético
especifico como es la translocación t(11;22)(q24;q12) (Figura
1 y 2), que supone una fusión génica de los genes
EWS y FLI1 y que se aprecia en más del 85% de los casos de
Es/PNET. En aproximadamente el 5% de los casos, el gen EWS está
implicado en otro tipo de translocaciones: t(21;22)(q12;q12) y t(7;22)(p22;q12)
que producen los genes de fusión EWS-ERG y EWS-ETV1 respectivamente
y que pueden ser importantes desde el punto de vista diagnóstico
y pronóstico (1).
Así, en nuestra experiencia, hemos descrito dos casos de
Es/PNET que presentaban junto a rasgos fenotípicos infrecuentes,
la t(2;22) que supone la fusión EWS/FEV y confiere una agresividad
biológica mayor que en aquellos casos de Es convencional
(2). Es importante pues, conocer la existencia de estas otras translocaciones,
especialmente en aquellos casos de Es que no presentan el típico
reordenamiento t(11;22).
Desde el punto de vista inmunohistoquímico, hemos demostrado
la expresión de la proteina FLI1 en el 100% de los casos
de Es (3). La expresión mas baja o la ausencia de expresión
de FLI1 en otros tumores de células redondas como neuroblastomas
o rabdomiosarcomas, revela a este marcador como útil junto
a la detección de CD99, para el diagnóstico diferencial
de estas neoplasias. Otros datos de importancia pronóstica
que hemos observado en Es/PNET hacen referencia a la deleción
homozigótica del locus génico 9p21 y a las mutaciones
puntuales del gen p53 (4). Estas alteraciones confieren mayor agresividad
biológica. Otro hecho interesante que hemos podido determinar
desde el punto de vista inmunohistoquimico, es la expresión
de c-KIT y su receptor SCF (Stem cell factor), en un alto porcentaje
de casos de Es/PNET (60%), lo cual abre posibilidades terapéuticas
alternativas con inmunoterapia frente a c-KIT (5). Otras moléculas
que podrían ser utilizadas como dianas terapéuticas
son el receptor del factor de crecimiento de la insulina (ILGF-R1),
FAS-FASL o c-erbB-2, que también se expresan en el Es/PNET
con un carácter más heterogéneo y variable.
Finalmente, el uso combinado de técnicas de hibridación
in situ fluorescente (FISH) e hibridación genómica
comparativa (CGH), nos ha permitido detectar alteraciones cromosómicas
secundarias como ganancias de los cromosomas 8 y 12, eventos probablemente
asociados a una ventaja proliferativa en Es/PNET(6).
El tumor desmoplásico de células redondas (DSRCT)
constituye una entidad particular con una diferenciación
divergente y una extremada agresividad. Afecta a niños y
jóvenes adultos siendo la localización más
frecuente la cavidad intraabdominal, formando grandes masas tumorales
que ocupan la pelvis y el retroperitoneo. Una de las principales
características de este tumor es su naturaleza plurifenotípica,
con expresión epitelial, neural y biogénica (1).
La histología muestra nidos o trabéculas de tamaño
y formas variables y delimitados por una lámina basal. Las
células son pequeñas, redondas, poligonales o en forma
de huso, estrechamente empaquetadas con bordes indistintos, y poseen
un citoplasma escaso que contiene una alta concentración
de glucógeno. Los núcleos son redondos o alongados,
e hipercromáticos (1).
Inmunohistoquímicamente presentan un patrón multifenotípico.
Las queratinas se expresan en todos los casos con un patrón
citoplasmático difuso. EMA se presenta en una gran proporción
de tumores con una distribución membranosa. La desmina y
la actina músculo lisa también se expresan en todos
los casos (Figura 1). Los marcadores neuroectodérmicos se
expresan en la mayoría de los casos, particularmente NSE,
PGP 9.5, y la sinaptofisina. Es menos frecuente la positividad para
CD99 y HNK-1 (1).
Citogenéticamente, los DSRCT están caracterizados
por la translocación reciproca t(11;22)(p13;q22) que resulta
en la fusión de los genes EWS y WT1 (7). Los cariotipos de
estos tumores son complejos con abundantes anomalías secundarias
que afectan posiblemente a muchos oncogenes y genes supresores de
tumores.
La familia de los tumores neuroblásticos (Neuroblastomas)
constituye el tipo tumoral sólido extracraneal más
frecuente diagnosticado en la infancia. Estos tumores, derivados
de la cresta neural y localizados en glándula suprarrenal
o en territorios extra-adrenales, presentan unas características
clínicas y biológicas curiosas, existiendo casos con
remisión espontánea, junto a otros con progresión
tumoral, escasa respuesta a la terapia y muerte del paciente. El
establecer por tanto criterios pronósticos es fundamental
para el manejo de los pacientes con neuroblastoma. Factores clínicos
como la edad del paciente (pronostico favorable en niños
menores de 1.5 años) y el estadio tumoral (favorable en estadios
localizados de la enfermedad), son criterios fundamentales para
la programación del tratamiento de los niños afectos
de neuroblastoma. Otros criterios con fuerte valor pronóstico
son biológicos y hacen referencia al contenido de ADN tumoral
con pronóstico favorable en aquellos tumores triploides,
en contraposición a los tumores desfavorables de contenido
diploide/tetraploide. Asimismo, la detección de amplificación
del oncogen MYCN, localizado en el cromosoma 2, y la perdida de
material genético o deleción 1p36 son otros factores
biológicos de valor pronóstico (8). Respecto a la
amplificación de MYCN, está demostrada su asociación
con los estadios avanzados de la enfermedad tumoral y a la falta
de respuesta a la quimioterapia. Sin embargo, la heterogeneidad
de estos tumores, queda reflejada en situaciones de valor pronóstico
desconocido como la ganancia de MYCN (equivalente a la detección
de 3-10 copias del gen), y la denominada amplificación focal,
que se manifiestan como nidos celulares sin amplificación
junto a focos de células con amplificación (9).
Además, sin recurrir a tecnología sofisticada y de
alto coste, el papel del diagnóstico histopatológico
en la tipificación y pronóstico del NB es cada vez
más relevante. Junto a la categorización básica
del tumor en subtipos de ganglioneuroma, ganglioneuroblastoma y
neuroblastoma, la clasificación de Shimada modificada (INPC),
recoge criterios morfológicos de valor pronóstico
como el grado de diferenciación, la riqueza de estroma schwaniano,
la evaluación del numero de figuras de mitosis y el índice
de cariorrexis (MKI), y la presencia de calcificaciones distróficas.
Basados en estos parámetros y en la edad del paciente se
delimitan dos grupos diferenciados con histopronóstico favorable
y desfavorable (10).
En nuestra experiencia, hemos confirmado en una amplia casuística
(209 casos), que la supervivencia global y el intervalo libre de
recaídas, están significativamente asociados a la
caracterización histopatológica favorable o desfavorable
siguiendo los criterios de la clasificación INPC (International
Neuroblastoma Pathology Classification) (11). Este último
resultado es especialmente interesante para aquellos casos de NB
localizado (estadios 1 y 2) en los que hemos demostrado, dentro
del contexto de un ensayo clínico cooperativo europeo, en
el que los 124 pacientes solo eran tratados quirúrgicamente,
que la histopatología es el factor pronóstico más
importante en el momento de predecir las recaídas y por tanto
el factor que influirá en la administración de quimioterapia
neoadyuvante en estos pacientes (12).
Los rabdomiosarcomas (RMS) constituyen los tumores de partes blandas
más frecuentes de la infancia (entre un 5% y un 8% de todas
las neoplasias malignas de la infancia). Desde el punto de vista
histológico se distinguen fundamentalmente los subtipos alveolar
y embrionario. El pronóstico depende del grado de extensión
de la enfermedad en el momento del diagnóstico y del tipo
histológico. Así, los pacientes con RMS alveolar presentan
una peor supervivencia que los pacientes con un RMS embrionario
(1).
Se han descrito numerosos anticuerpos frente a filamentos proteínicos
citoplasmáticos: desmina, actina músculo específica
y miosina sarcomérica; y frente a proteínas no filamentosas
del citoplasma: mioglobina, creatinina-quinasa M, distrofina, titina
y troponina (Figura 1). Las proteínas nucleares MyoD1 y miogenina
son muy específicas es estos tumores. La desmina se expresa
en el 95% de los RMS, siendo el marcador más útil.
Los RMS embrionarios expresan miogenina y MyoD1 en aproximadamente
el 90% de los casos. También se ha descrito la expresión
de otros marcadores menos específicos como son, el HNK-1,
CD99, NSE y la CAM 5.2 (1).
Citogenéticamente, los RMS alveolares y embrionarios constituyen
dos entidades distintas. Los RMS embrionarios no muestran ninguna
reordenación cromosómica específica, sin embargo,
la mayoría de los tumores son hiperdiploides con un incremento
en el número de copias para los cromosomas 2, 7, 8, 12 y
13 en particular. La hibridación genómica comparativa
(CGH) confirma estos hallazgos, mostrando ganancias de cromosomas
enteros, como por caso los cromosomas 2, 13, 12, 8, y 7 entre el
50-60% de los casos y de los cromosomas 17, 18 y 19 en el 40% de
los tumores. También han sido descritas las pérdidas
de los cromosomas 16 y 10 y 15 y 15 en el 30%-40% y 20% de los casos
respectivamente (13).
Los estudios de polimorfismos muestran que los RMS están
asociados con la pérdida de heterozigosidad en el locus 11p13.
Por el contrario, los RMS alveolares se caracterizan por dos translocaciones
específicas: t(2;13)(q35;q14) y t(1;13)(p36;q14), que se
encuentran en el 80% y 15% de los casos respectivamente. Resultado
de estas translocaciones son los genes de fusión PAX3-FKHR
en el caso de la t(2;13) y PAX7-FKHR para la t(1;13). También
se han descrito amplificaciones en forma de dobles minutos y los
estudios con CGH han demostrado la presencia de amplificaciones
génicas en los amplicones 12q13-15 (50% de los casos), 2p24
(36%), 13q14, 13q32 y 1q36 (14% de los casos), 1q21 y 8q13-q21 (7%).
Es especialmente interesante el amplicón 12q13-15 en el que
se encuentran localizados los genes CHP, MDM2, CDK4 y SAS, importantes
reguladores del ciclo celular. Los casos con amplificación
del locus 2q24 se asocian a la amplificación del gen MYCN,
pero a diferencia de los neuroblastomas, no se ha encontrado ninguna
asociación con el pronóstico de los pacientes con
un RMS alveolar. Por otra parte, los casos con la fusión
PAX7-FKHR a menudo muestran una amplificación de este gen
quimérico. Parece evidente que los RMS alveolares y embrionarios
constituyen dos entidades con características citogenéticas
y moleculares diferentes, y por tanto, la caracterización
de dichas diferencias tiene un indudable valor, no solo desde el
punto de vista diagnóstico, si no también pronóstico
(14).
El condrosarcoma mixoide extraesquelético (CME) es un tumor
maligno de partes blandas y distinto del condrosarcoma esquelético
primario con alteración mixoide. Constituye una entidad rara
con una incidencia del 2.3% de todos los sarcomas de partes blandas,
siendo los hombres los que lo presentan con una mayor frecuencia
a razón de 2:1 con respecto a las mujeres. En el 75% de los
casos se localizan en las extremidades inferiores. Desde el punto
de vista histológico el CME, está compuesto por células
redondas o ligeramente elongadas con características parecidas
a los condroblastos (Figura 1). El diagnóstico histológico
puede entrañar dificultades, especialmente en las formas
con alta densidad celular desprovista de matriz mixoide (1). En
este sentido, la citogenética y la biología molecular
constituyen una importante herramienta. Más del 75% de los
CME presentan la translocación t(9;22)(q22;q12) que como
resultado produce el gen de fusión EWS-TEC(CHN) que se puede
identificar por medio de RT-PCR sobre material fijado e incluido
en parafina. Recientemente se han descrito otras dos translocaciones:
t(9;17)(q22;q11) y t(9;15)(q22;q21) donde los genes de fusión
RBP56-TEC y TCF12-TEC se encuentran implicados respectivamente (15).
Los sarcomas sinoviales (SS) son neoplasias que acontecen entre
un 5-8% del total de tumores de partes blandas. El 90% de los casos
se presenta en articulaciones y tendones de niños o jóvenes
adultos, más frecuentemente en sexo masculino. Se han descrito
las variantes histológicas bifásica y monofásica.
En la variante bifásica se distingue un componente epitelial
que expresa marcadores epitelioides (pan-keratina, AE1-AE3, CAM
5.2) y EMA en casi el 100% de los casos, así como S100, CEA,
CD99 y menos frecuentemente CD56 y PGP9.5. Por su parte en el SS
monofásico, las pequeñas células fusiformes
expresan vimentina (Figura 1), pero ningún marcador epitelial,
exceptuando algunas células aisladas que puedan expresar
citoqueratinas y EMA (1, 16).
Desde el punto de vista citogenético se caracterizan por
la translocación específica t(X;18)(p11.2;q11.2) presente
en más del 90% de los sarcomas sinoviales, y que en más
de un tercio se presenta como la única alteración
citogenética. Esta translocación resulta en la producción
de dos genes de fusión en los cuales la secuencia 5' del
gen SYT (18q11.2) se une a la secuencia 3' de dos genes intimamente
relacionados en la posición Xp11.2, designados como SSX1
y SSX2. Los genes SSX1 y SSX2 probablemente derivan de un proceso
de duplicación en el proceso evolutivo lo cual explica el
alto porcentaje de homología entre ambas proteínas
(81%). Como se ha demostrado en estudios recientes, la identificación
de los diferentes tipos de fusión en los SS por medio de
RT-PCR, constituye una herramienta valiosa en el diagnóstico
diferencial de estos tumores (16) (Figura 2). También, estos
últimos años se ha demostrado que los receptores c-KIT
y PDGFRA juegan un importante papel en el crecimiento del tumor
y en la progresión de los sarcomas sinoviales en los que
la expresión de c-KIT se ha descrito en un 70% de los casos
(17). En este contexto, se ha sugerido que los casos positivos para
c-KIT se pudieran beneficiar de terapias con inhibidores específicos
de receptores tirosina quinasa, como es el STI-571 (Glivec, Novartis)
en los tumores del estroma gastrointestinal (GIST). Sin embargo,
los estudios en fase II no están siendo muy prometedores.
A diferencia de los GIST, no se han descrito mutaciones en los genes
c-KIT y PDGFRA de los SS (18).
Resulta por tanto evidente que como resultado de los nuevos avances
en las tecnologías de genética y proteómica
para el conocimiento de los mecanismos moleculares de los sarcomas
de células redondas, los patólogos deberán
conocer las nuevas tecnologías en biología molecular
aplicable al diagnóstico de los tejidos y células
junto con los fundamentos en la biología molecular del tumor
y su utilidad clínica.
Arriba
REFERENCIAS
- Peydro-Olaya A, Llombart-Bosch A, Carda-Batalla C, Lopez-Guerrero
JA. Electron microscopy and other ancillary techniques in the
diagnosis of small round cell tumors. Semin Diagn Pathol, 20(1):25-45,
2003.
- Navarro S, Noguera R, Pellin A, Lopez-Guerrero JA, Rosello-Sastre
E, Cremades A, Llombart-Bosch A. Atypical pleomorphic exotraosseous
Ewing tumor/peripheral primitive neuroectodermal tumor with unusual
phenotypic/genotypic profile. Diagn Mol Pathol 11(1):9-15, 2002
- Llombart-Bosch A, Navarro S. Immunohistochemical detection of
EWS and FLI-1 proteins in Ewing sarcoma and primitive neuroectodermal
tumors: comparative analysis with CD99 (MIC-2) expresión.
Appl Immuno Mol Morphol 9 (3):255-260, 2001.
- Lopez-Guerrero JA, Pellin A, Noguera R, Carda C, Llombart-Bosch
A. Molecular analysis of the 9p21 locus and p53 genes in Ewing
family tumors. Lab Ivest 81(6):803-814, 2001.
- Navarro S, Llombart-Bosch A, Giraudo P, Smirnov A, Petrovichev
N, Alvarado I. Immunophenotypic profile of anti-apoptotic and
neuroectodermal differentiation pathways in the Ewing’s family
of tumors (EFT). Histopathology, 2004, in press.
- Llombart-Bosch A, Ferrer J, Noguera R, Lopez-Guerrero JA, Ballester
S, Carda C, Pellin A. Sequential cytogenetic numerical changes
in Ewing’s sarcoma: a xenotransplantation model. Lab Invest
84 (Suppl. 1): 17A, 2004.
- Ladanyi M, Gerald W. Fusion of the EWS and WT1 genes in the
desmoplastic small round cell tumor. Cancer Res 54:2837-40, 1994.
- Brodeur GM. Neuroblastoma: biological insights into a clinical
enigma. Nature Rev Cancer, 3:203-216, 2003.
- Noguera R, Cañete A, Pellin A, Ruiz A, Tasso M, Navarro
S, Castel V, Llombart-Bosch A. MYCN gain and MYCN amplification
in a stage 4s neuroblastoma. Cancer genet cytogenet 140:157-161,
2003.
- Shimada H, Ambros I, Dehner LP. Et al The international Neuroblastoma
Pathology Classification (the Shimada system). Cancer, 86:364-372,
1999.
- Burgues-Gasion O. Valor histopronostico de la clasificacion
histopatológica (INPC) en el neuroblastoma infantil. Estudio
comparativo con otros factores pronosticos. Tesis doctoral. Universidad
de Valencia. Julio 2003.
- Navarro S. Classificacion Internationale des Neuroblastomes.
Groupe d’etude Europeen du Neuroblastome. Communaute Europeenne.
LNESG1. Paris, 13 November 2003.
- Weber-Hall S, Anderson J, McManus A, Abe S, Nojima T, Pinkerton
R, Pritchard-Jones K, Shipley J. Gains, losses, and amplification
of genomic material in rhabdomyosarcoma analyzed by comparative
genomic hybridization. Cancer Res 56:3220-3224, 1996.
- Gil-Benso R, Lopez-Gines C, Carda C, Lopez-Guerrero JA, Ferrer
J, Pellin-Perez A, Llombart-Bosch A. Cytogenetic and molecular
findings related to rhabdomyosarcoma. An analysis of seven cases.
Cancer Genet Cytogenet 144:125-133, 2003.
- Rubin BP, Fletcher JA. Skeletal and extraskeletal myxoid chondrosarcoma:
related or distinct tumors? Adv Anat Pathol 6: 204-212, 1999.
- Llombart-Bosch A, Lopez-Guerrero JA, Peydro-Olaya,A. Synovial
sarcoma (SS): new perspectives supported by modern technology.
Arkh Patol 64:39-47, 2002.
- Smithey BE, Pappo AS, Hill DA. C-kit expression in pediatric
solid tumors: a comparative immunohistochemical study. Am J Surg
Pathol 26:486-492, 2002.
- A. Llombart-Bosch, J. A. López-Guerrero, S. Navarro,
R. Noguera, C. Carda, A. Pellín. Molecular analysis of
the c-KIT and PDGFRa genes in a series of molecularly well characterized
synovial sarcomas. Lab Invest 84 (Suppl. 1): 17A, 2004.
Arriba
Figura 1
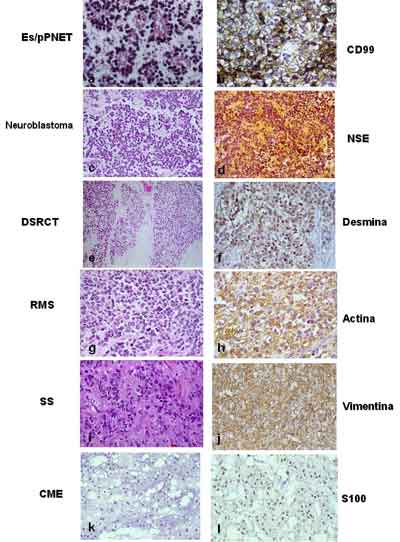
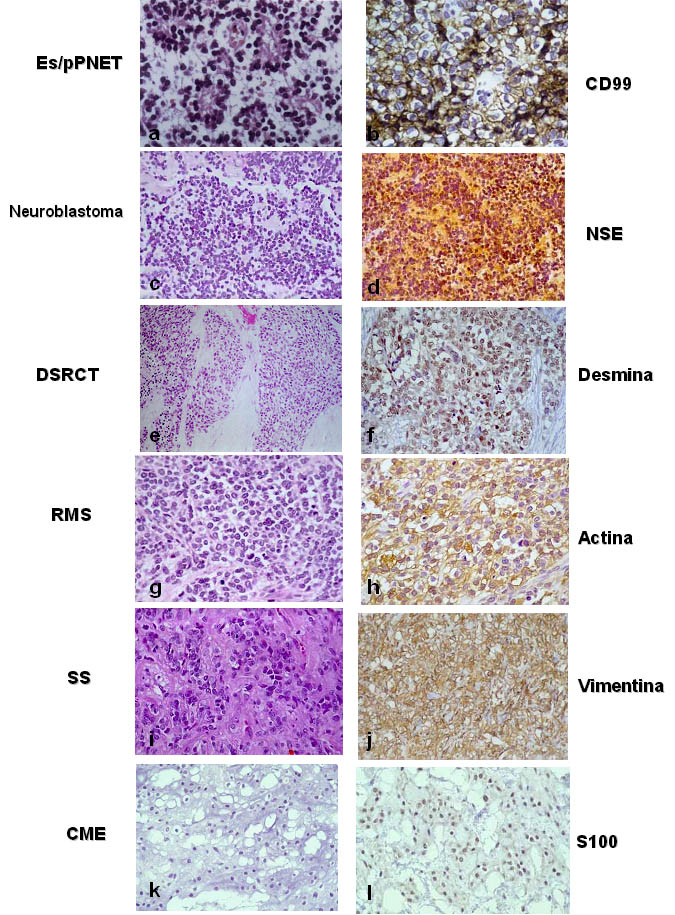
Figura 1. Aspectos histológicos e inmunohistoquímicos
de los tumores de células redondas: a y b, estructuras de
tipo pseudorosetas en un pPNET y tinción membranosa característica
para CD99; c y d, neuroblastoma pobremente diferenciado con expresión
de enolasa (NSE); e y f, tumor desmoplásico de células
redondas con la clásica inmunotinción para desmina;
g y h, rabdomiosarcoma sólido alveolar con expresión
citoplásmica en actina; i y j; sarcoma sinovial monofásico
con expresión de vimentina; k y l, condrosarcoma extraesquelético
con típica inmuinotinción nuclear para S100.
Arriba
Figura 2
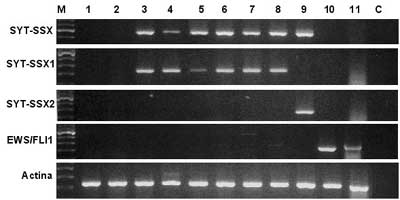
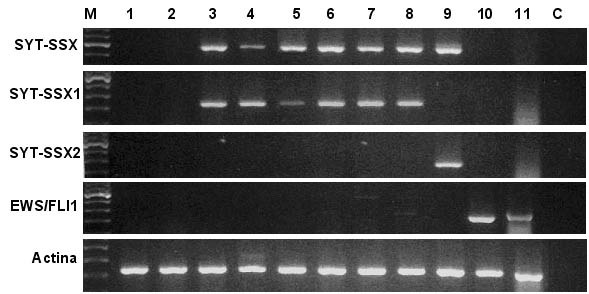
Figura 2. Geles representativos de electrofoeresis
en los que se muestra el resultado de las reacciones de RT-PCR con
iniciadores específicos para los transcritos de fusión
SYT-SSX1, SYT-SSX2 y EWS-FLI1. Pocillos: 1y 2, muestras de cáncer
de mama; 3-8, sarcomas sinoviales con la fusión SYT-SSX1;
9, sarcoma sinovial con la fusión SYT-SSX2; 10-11, sarcomas
de Ewing con la fusión EWS-FLI1; C, control de reactivos.
Arriba
--------------------------------------------------------------------------------
|
|
|