| Información || Congresos || Cursos || Territoriales || Noticias || Patología || Telepatología |
Galdakao (Vizcaya). País Vasco
HOSPITAL SANTIAGO APÓSTOL. Vitoria-Gasteiz
J. De Diego, B. Catón, N.Arbide, N.Saracibar (Sº de Anatomía Patológica)
AMILOIDOSIS PRIMARIA ASOCIADA A ENFERMEDAD POR DEPÓSITO DE INMUNOGLOBULINAS.
RESUMEN DE HISTORIA CLÍNICA
Varón de 70 años, con síndrome nefrótico, gammapatía monoclonal en suero y orina tipo lambda, serie ósea normal, células plasmáticas en médula ósea inferiores al 10% sin plasmocitosis periférica y extenso depósito sistémico de amiloide.
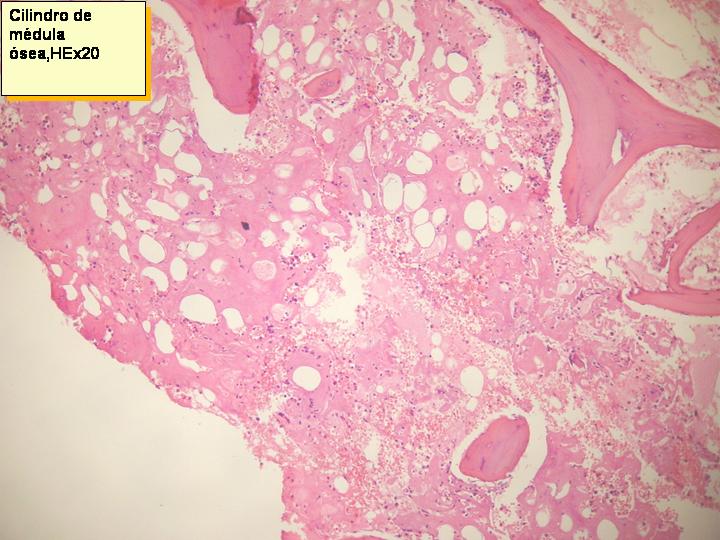
Cilindro de Médula ósea. HE x20

Hospital Santiago Apóstol de Vitoria-Gasteiz
DIAGNÓSTICO
AMILOIDOSIS PRIMARIA ASOCIADA A ENFERMEDAD POR DEPÓSITO DE INMUNOGLOBULINAS.
COMENTARIOS
La amiloidosis es un grupo heterogéneo de enfermedades asociadas al depósito extracelular de una proteína fibrilar anómala denominada amiloide. La amiloidosis puede ser primaria (AL), secundaria (AA) y hereditaria y los depósitos pueden ser localizados o sistémicos. El amiloide se define como una sustancia proteinácea, patológica, hialina y eosinófila que se deposita entre las células de los órganos y tejidos y provoca la atrofia celular. En un 95% se encuentra constituido por largas fibrillas, no ramificadas, con estructura beta plegada responsable de la birrefringencia verde manzana cuando se estudia mediante tinción con Rojo Congo y observación con luz polarizada. El 5% restante se encuentra constituido por una glucoproteina, denominada componente P, responsable de la afinidad del depósito tisular del amiloide.
Se han identificado hasta 15 formas bioquímicas distintas de amiloide y de acuerdo a su composición las amiloidosis se clasifican en:
1. Primarias: amiloide AL, constituido por cadenas ligeras de inmunoglobulinas monoclonales y asociadas a displasias de células plasmáticas.
2. Secundarias: amiloide tipo AA que es una proteína sintetizada en el hígado con relación a procesos inflamatorios crónicos.
3. Formas familiares (Fiebre mediterránea familiar, amiloide tipo AA) o hereditaria (polineuropatías amiloidóticas familiares con depósito de transtirretina mutante).
4. Amiloidosis de la hemodiálisis con depósito de beta-2 microglobulina.
La amiloidosis primaria sistémica es una enfermedad poco frecuente, con una incidencia estimada de 8 casos/millón al año y con una prevalencia constante. Para su diagnóstico se requieren una constelación de datos clínicos y patológicos, siendo imprescindible la demostración de un componente monoclonal de inmunoglobulinas en suero y orina. Es el denominado componente M y estaría relacionado con algún tipo de discrasia de células plasmáticas.
La O.M.S. reconoce cinco tipos principales de neoplasias de células plasmáticas:
1. Mieloma de células plasmáticas clásico con criterios definidos y sus variantes (mieloma no secretor, mieloma indolente, mieloma Smoldering y leucemia de células plasmáticas), así como lesiones precursoras (gammapatía monoclonal de significado indeterminado-MGUS).
2. Plasmocitoma solitario de hueso y plasmocitoma extramedular.
3. Enfermedad por depósito de inmunoglobulinas: amiloidosis primaria y enfermedad sistémica por depósito de cadenas ligeras y pesadas.
4. Mieloma osteoesclerótico o síndrome de POEMS.
5. Enfermedad de cadenas pesadas (HCD).
Las enfermedades por depósito de inmunoglobulinas monoclonales forman parte del espectro del mieloma múltiple aunque no cumplen los criterios establecidos para mieloma, en el momento del diagnóstico. La diferencia fundamental entre la amiloidosis primaria y la enfermedad por depósito por cadenas ligeras estriba en la naturaleza diferente del amiloide. Mientras que la amiloidosis primaria el depósito está constituido por cadenas ligeras de inmunoglobulinas tipo lambda con estructura plegada en beta y birrefringencia con Rojo Congo y componente P del amiloide, en la enfermedad por depósito de cadenas ligeras y pesadas el depósito se encuentra constituido por cadenas ligeras tipo Kappa que se depositan en forma de material amorfo no plegado en beta (Rojo Congo negativo) y sin componente P.
En el tratamiento de la amiloidosis primaria se utilizan agentes quimioterápicos y trasplante autólogo de células madre hemopoyéticas. Aún con tratamiento la supervivencia media estimada es de dos años desde el momento del diagnóstico y la muerte sobreviene por fallo de los órganos y sistemas afectados por el depósito de amiloide (corazón, riñón, hígado, nervios periféricos).
BIBLIOGRAFÍA
-Ramzi S. Cotran, Vinay Kumar and Tucker Collins. Patologia estructural y funcional, Ed. McGraw Hill-Interamericana, ( 6ª edición) , 267-273.
-World Health Organization Classification of Tumors. Tumors of Haematopoietic and Lymphoid Tissues. IARC Press, 142, 156.
-Gertz MA, Rajkumar SV. Primary sistemic amyloidosis. Current treatment options in Oncology. 2002, Jun, 3(3): 261-71.
-Kyle RA, Bayrd ED. Amyloidosis: review of 236 cases, Medicine (Baltimore), 1975 Jun; 54 (4): 271-99.
-Kyle RA, Lines A, Beard CM et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 Through 1989, Blood, 1993 Jan 15; 81 (2): 564-5.
-Strege RJ, Saeger W, Linke RP. Diagnosis and Immunohistochemical classification of sistemic amyloidosis. Report of 43 cases in an unselected autopsy series. Virchows Arch, 1998 Jul; 433 (1): 19-27.
[Caso anterior] [Página Inicio de la Reunión Territorial] [Siguiente caso]