| Información || Congresos || Cursos || Territoriales || Noticias || Patología || Telepatología |
Galdakao (Vizcaya). País Vasco
HOSPITAL GENERAL YAGÜE. Burgos
Angeles Baños Baños, Angel Velasco Osés y Enrique García Toro
SINDROME DE BUDD-CHIARI
RESUMEN DE HISTORIA CLÍNICA
Varón de 43 años que ingresa 8 días antes de su fallecimiento con insuficiencia hepática e hipertensión portal. Se detecta un gran quiste hidatídico. Se sospecha cirrosis hepática de fondo o compromiso vascular, con gran ascitis recidivante o una etiología tóxica o por setas.
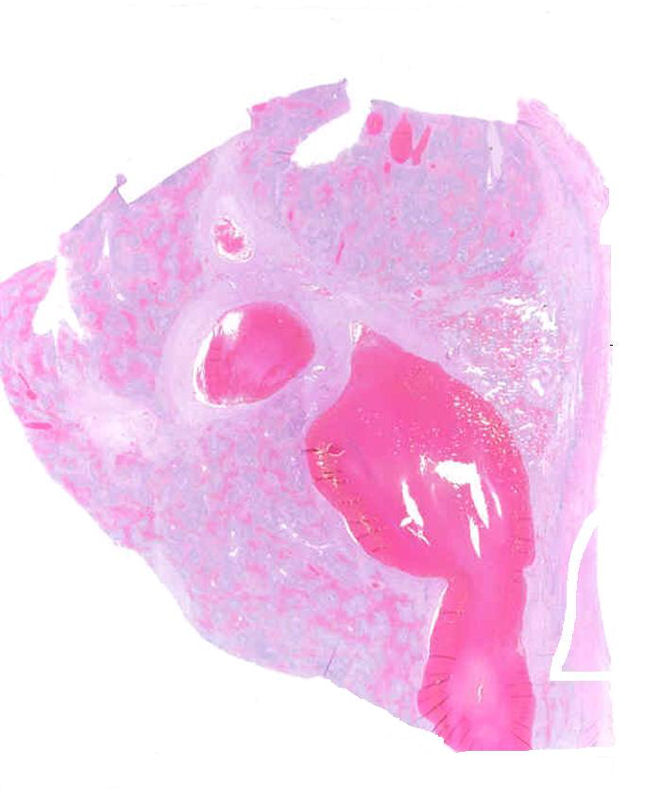
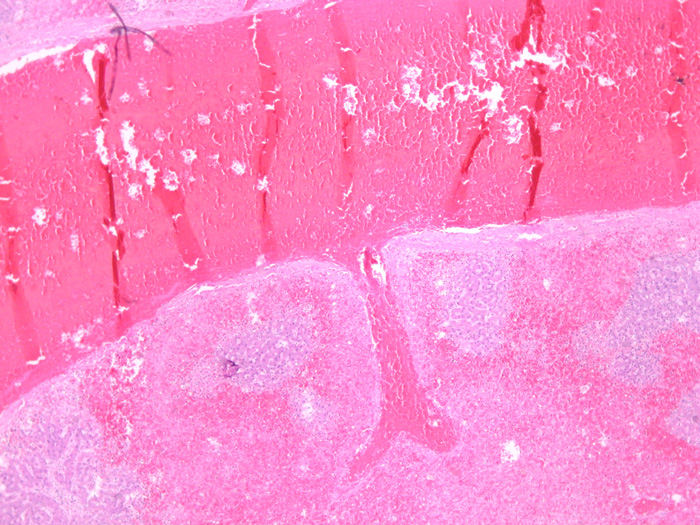
DIAGNÓSTICO
SINDROME DE BUDD-CHIARI
COMENTARIOS
El síndrome de Budd-Chiari consiste en la oclusión del drenaje venoso del hígado ya sea por el compromiso total o parcial de las venas suprahepáticas(derecha, media e izquierda) o del segmento distal de la vena cava inferior.
Fué descrito por primera vez en 1845 por Budd y posteriormente Chiari en 1899, hace una detallada descripción clínico-patológica de 13 casos, varios de ellos de endoflebitis con posterior trombosis
El Síndrome de Budd-Chiari presenta un 40% de casos de etiología desconocida, un 25% por trastornos hematológicos y un 10% de origen infeccioso (esquistosiomasis, abscesos amebianos) en Occidente. En Asia y Africa el 40% se debe a la existencia de lesiones congénitas en vena cava inferior (atresia venosa, presencia de diafragmas o membranas). Los desórdenes hematológicos están relacionados con un estado de hipercoagulabilidad: policicitemia vera, hemoglobinuria paroxística nocturna, sd. Antifosfolípido, anemia de células falciformes, procesos mieloproliferativos, déficit hereditarios de factores de coagulación( antitrombina III, Factor V de Leiden, Proteina C y Proteina S), uso crónico de anticonceptivos, Otras causas incluye: embarazo y estado posparto, tumores endovenosos de la cava inferior como el angiomioleioma, neoplasias locales o metástasis ganglionares que comprometan la unión de las venas hepáticas con la Cava inferior, traumatismos abdominales y uso a altas dosis de quimioterápicos en el transplante autólogo o alogénico de médula ósea.
El síndrome de Budd-Chiari puede cursar de forma fulminante, aguda o crónica, en función del grado y de la velocidad de la obstrucción vascular. La forma aguda, es la forma de presentación clínica más frecuente en la actualidad.
El hallazgo macroscópico en los cuadros de hepatopatía fulminante es el de dos patrones fundamentales:
-Patrón difuso: disminución del tamaño hepático, con afectación uniforme de todo el órgano que toma un color amarillento atrofia aguda amarillenta del hígado o hígado aumentado de tamaño, superficie lisa de color rojo con áreas moteadas amarillentas que indican pérdida de células perivenulares confluentes y congestión, con escasez de hepatocitos periportales.
En algunas ocasiones se ha encontrado cierta relación entre estos hallazgos y la causa: hepatitis tóxica por paracetamol, tetracloruro de carbono o envenenamiento por Amanita phalloides
-Patrón mapeado: áreas de colapso reticulínico total del parénquima alternando con zonas donde aparentemente ha desaparecido el parénquima y aparecen grandes nódulos de regeneración. Este patrón habitualmente se asocia a una patología hepática de origen vírico de curso más prolongado.
Microscópicamente en el caso del Síndrome de Budd-Chiari, la obstrucción puede verificarse en cualquier punto de las venas suprahepáticas o en la cava inferior y hasta en un 20% de los casos se asociara a una secundaria trombosis de la vena porta.
En los procesos agudos, histológicamente no diferirá del hígado de shock. Presentando dilatación sinusoidal, éstasis venoso, reacción inflamatoria celular y necrosis hemorrágica de hepatocitos de la zona 3, cuya intensidad dependerá del grado y rapidez de la obstrucción venosa. Lógicamente se añade la trombosis de las venas centrolobulillares que diferencian al sindrome .
En procesos crónicos , se verificará fibrosis y nódulos de regeneración con la consecuente pérdida de la normal arquitectura hepática. La hipertensión portal puede causar esplenomegalia y circulación colateral porto-sistémica
BIBLIOGRAFÍA
ANDERSON´S PATHOLOGY. Pg: 1415-1416. 10º Ed. Ivan Damjonov. y James Linder.
M.A. LÓPEZ GARRIDO Y M.E. CERVILLA SAEZ DE TEJADA: quiste hidatídico como causa de Budd-Chiari. Hospital Universitario de Granada
ALBRECHT KRÄMER, FRANCISCO VALDÉS, FRANCISCO CRUZ, SERGIO GONZALEZ.: Síndrome de Budd-Chiari, tratamiento quirúrgico de 2 casos. Pontificia Universidad Católica de Chile
PONIACHIK, QUERA, LUI: Insuficiencia hepática fulminante. Rev-Med_Chil.2002 Jun; 130(6): 691-8
GARCIA-PAGAN, PERELLO, BOSCH: Síndrome de Budd-Chiari. Gastroenterología-Hepatolo. 2000 Dec; 23(10): 491-7
KHUROO MS, DATTA DV, KHOSHY A, MITRA SK, CHHUTTANI PN. Alveolar hydatid disease of the liver with Budd-Chiari syndrome. Postgrad Med J. 1980 Mar;56(653):197-201
MARTIN HERRERA L, CHAMORRO LOPEZ J, BASCUNANA QUIRELL A. Budd-Chiari syndrome caused by hepatic hydatid cyst. Med Clin (Barc). 1982 Jan 25;78(2):75-6.
ROBOTTI GC, MEISTER F, SCHRODER R.. Budd-Chiari syndrome in liver echinococcosis. Rofo. 1985 May;142(5):511-3.
[Caso anterior] [Página Inicio de la Reunión Territorial] [Siguiente caso]