| Información || Congresos || Cursos || Territoriales || Noticias || Patología || Telepatología |
Leioa, 4 de noviembre de 2005
HOSPITAL DE GALDAKAO.
Dr. Eduardo de Miguel Herrán, Dra. Beatriz Eizaguirre Zarza, Dr. Alberto Saiz López, Dra. Igone Imaz Murga, Dr. José Antonio Alvarez Martínez, Dra. Juana Rodríguez González, Dra. Pilar Manrique Martínez (*), Dr. Koldo Atutxa Aresti (**) y Dr. Iñaki Zabalza Estévez.
* Servicio de Dermatología. Hospital de Galdakao. ** Servicio de Hematología. Hospital de Galdakao.MASTOCITOSIS SISTÉMICA
RESUMEN DE HISTORIA CLÍNICA
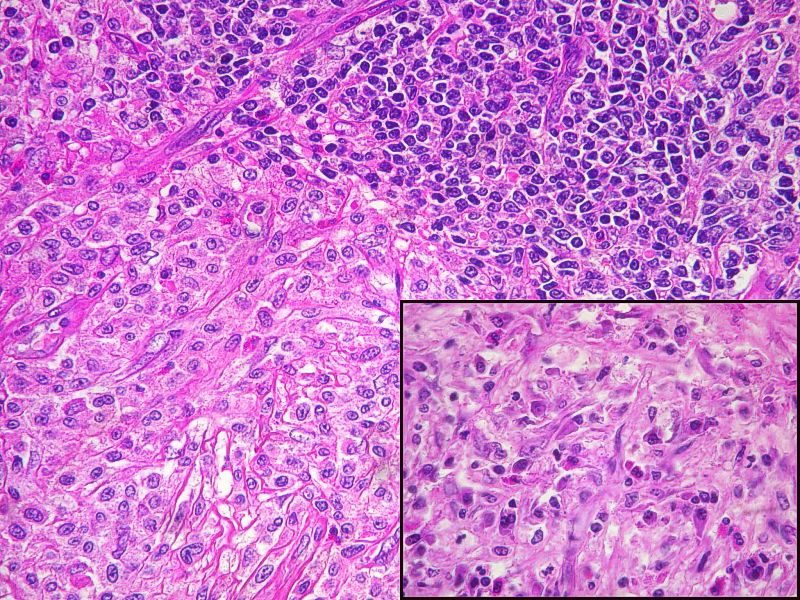
Caso 1: Varón de 45 años con lesión polipoide en muslo de 1,2 cms, remitida como acrocordón.
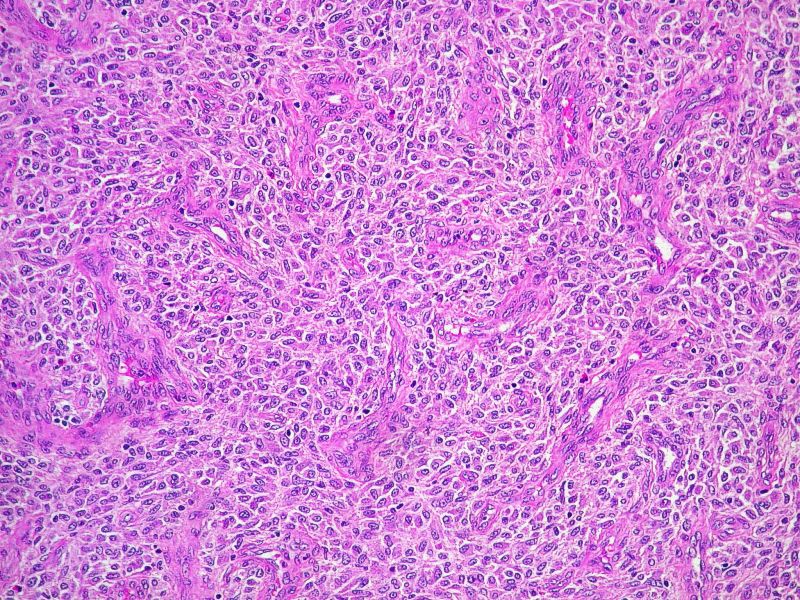
Caso 2: varón de 61 años controlado por el Servicio de Hematología por síndrome mielodisplásico de larga evolución, al que se le efectúa una esplenectomía de la cual os remitimos un corte
c-kit

Triptasa
DIAGNÓSTICO
MASTOCITOSIS SISTÉMICA
COMENTARIO
Presentamos dos casos de mastocitosis sistémica, que en parte se complementan. El primer caso corresponde a un varón de 45 años que presentaba lesión polipoide en muslo, remitido clínicamente como acrocordón, en el segundo caso correspondiente a un varón de 61 años con síndrome mielodisplásico conocido de larga evolución, que presenta un cuadro adenopático abdominal y esplenomegalia.
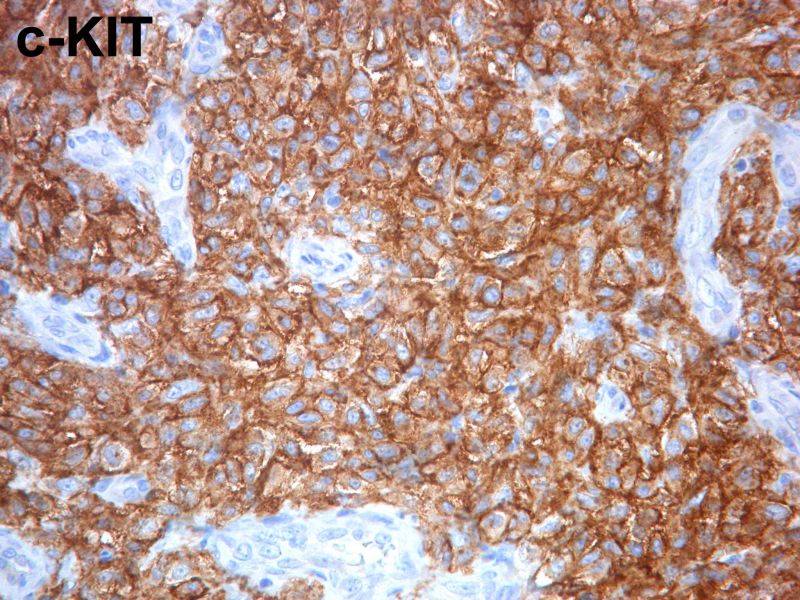
En el primer caso se objetivó una forma atípica de infiltración cutánea por células mastocitarias, morfológicamente difíciles de reconocer como tal, si bien resultaron ser intensamente positivas para técnicas especiales de Triptasa, C-kit y CD-25.
En el segundo caso la pieza de esplenectomia que se nos remitió mostraba un infiltrado parcheado asociado a bandas de fibrosis de disposición parafolicular en la teórica zona marginal de la pulpa blanca, pero también de forma aleatoria en localización perivascular y paratrabecular. El infiltrado estaba constituido por células mononucleadas de apariencia monomorfa y citoplasmas pálidos, distribuidas preferentemente en torno a folículos, asociandose fenómenos de fibrosis intensa y un cortejo inflamatorio acompañante rico en eosinófilos, linfocitos, células plasmáticas y fibroblastos. Dichos agregados celulares resultaron ser intensamente positivos para técnicas especiales de Triptasa, C-kit y CD-25. Con el diagnóstico de mastocitosis esplénica se hizo un estudio en suero del enfermo para Triptasa que se encontraba notablemente elevada, estudio óseo por resonancia en el que se identificaba una hipo-intensidad de señal difusa a nivel fundamentalmente de cuerpos vertebrales y se repitió de nuevo estudio de médula ósea, en la que aparte de los cambios mielodisplásicos, no se objetivaban signos de infiltración mastocitaria. Dada la peculiar presentación en este segundo caso de una mastocitosis sistémica con afectación aparentemente exclusivamente en bazo, sin lesiones cutáneas asociadas, ni evidencia de afectación de médula ósea, se hizo estudio genético, resultando positivo y estudio de citometria en médula ósea observándose únicamente un 0,02 % de mastocitos, todos ellos de inmunofenotipo aberrante, por lo que se hizo un diagnóstico en base al inmunofenotipo de compatibilidad con infiltración de médula ósea por mastocitosis sistémica.
La clasificación de consenso de las mastocitosis tras la Reunión de Trabajo en Viena del año 2000, se encuentra recogida en el libro de la WHO, dividiéndolas fundamentalmente en dos grandes grupos, el más frecuente va a corresponder a las formas cutáneas (al menos el 80% de los casos), con sus diferentes variantes, fundamentalmente en la forma de urticaria pigmentosa, afectando fundamentalmente a niños en edades tempranas de la vida, con muchos casos de autoresolución y en general buen pronóstico (el enfermo refiere que en la infancia presentó lesiones de iguales características que se resolvieron espontáneamente). El resto de las formas van a corresponder a formas sistémicas, diagnosticadas en su inmensa mayoría a través de cilindros de médula ósea, siendo típica la asociación a lesiones cutáneas, en al menos el 50% de los casos, dividiéndolas en diferentes variantes, la más frecuente la forma indolente, la segunda la asociada a desordenes clonales mieloides de estirpe no mastocitaria (aproximadamente un 20% de los casos), en tercer lugar las formas agresivas y resultan excepcionales las formas de leucemia y sarcoma de células mastocitarias, así como el mastocitoma extracutáneo. En nuestro caso, el primer caso presentado de extensa afectación cutánea y médula ósea, encajaría dentro de una forma indolente de mastocitosis sistémica, si bien dada la gran afectación medular (superior al 30%) y extensa afectación cutánea, quizás al enfermo se de debería de clasificar en la subvariante provisional Smouldering. El segundo caso, por corresponder a una mastocitosis sistémica , histológicamente confirmada en bazo, asociada a síndrome mielodisplásico, con afectación ósea por técnicas de imagen y medular por técnicas de citometria de flujo, encajarian tanto dentro de una forma asociada a desordenes clonales medulares de estirpe no mastocitaria como a una forma de mastocitosis sistémica agresiva, por lo tanto de bastante peor pronóstico que el caso número 1.
Decir también que existen unos criterios de consenso diagnósticos de mastocitosis sistémica, basados fundamentalmente en la presencia de agregados multifocales celulares en médula ósea u otros sitios extracutáneos, en los que existan quince o más mastocitos confirmados por técnicas especiales de Triptasa u otras técnicas de inmunohistoquímica (criterio mayor), así como criterios menores, como son en primer lugar la presencia de más de un 25% de mastocitos de contornos fusocelulares o signos de atipia e inmadurez en extendidos citológicos de médula ósea, en segundo lugar el estudio genético positivo, en tercer lugar la coexpresión CD-117 con CD-2 y/o CD-25, y en cuarto lugar los niveles de Triptasa superior a 20 mgr/ml, excluidos aquellos casos que asocien una neoplasia medular clonal de otra estirpe diferente a la mastocitaria. La suma de un criterio mayor y uno menor o de tres criterios menores, nos dan el diagnóstico.
Decir también que la máxima rentatibilidad diagnóstica se obtiene con biopsias de médula ósea, ya que nos van a permitir ver los agregados celulares, así como su morfología fusocelular o no, así como la coexpresión de CD-117 con CD-2 y CD-25, que nos permitirían efectuar un diagnóstico de mastocitosis sistémica independientemente de otros hallazgos.
Desde un punto de vista inmunohistoquímico resultan decisivas las técnicas de Triptasa, si bien estas nos van a teñir tanto células tumorales como mastocitos (hiperplásicos), así como fundamentalmente las técnicas especiales de CD-2 y CD-25, las cuales únicamente resultan positivas en mastocitos de carácter neoplásico.
Decir también que en cuanto al tratamiento aparte del sintomático y PUBA, en las formas más agresivas el tratamiento de elección es el Interferon- 2- alfa, asociado no a glucocorticoides, con un índice de respuestas de un 21%. En caso de ineficacia o resistencia a dicho tratamiento, y basándose en estudio genéticos moleculares de los enfermos, se dividirían a estos en dos grandes grupos, el primero y más numeroso en el que se observa la típica mutación de c-Kit en el codón 816, así como aquellos casos no aclarados molecularmente, se beneficiarían del tratamiento con Cladribine, reservándose el Imatinib para aquellos casos en los que se observe la fusión genética de FIP-1 L-1 PDGFRA (muy típico de los síndromes hipereosinofílicos, así como de las mastocitosis sistémicas con eosinofilia asociada), con resultados prometedores en ambos casos.
BIBLIOGRAFÍA
1.- Tumours of haematopoietic and lymphoid tissues
World health organization classification of tumours.
2.- Diagnostic criteria an classification of mastocytosis: a consensus proposal.
Leuk Res. 2001 Jul; 25(7): 603-25
3.- CHIC 2 Deletion, a surrogate for FIP1L1-PDGFRA fusion
Blood, 1 November 2003, Vol. 102, nª 9, pp 3093-3096
4.- Mastocytosis: Current concepts in diagnosis and treatment
Ann Hematol (2002) 81: 677-690.
5.- Splenic mastocytosis: report of two cases and delection of the transforming somatic c-KIT mutation D816V.
Leuk lymphoma 2004. Apr 45 (4): 723-9
[Caso anterior] [Página Inicio de la Reunión Territorial] [Siguiente caso]