|
Información || Congresos || Cursos || Territoriales || Noticias || Patología || Telepatología |
|
Información || Congresos || Cursos || Territoriales || Noticias || Patología || Telepatología |
| . |
[Noticias]
[Boletín de la SEAP] [Bolsa
de Trabajo]
[Convocatoria] [Programa] [Hospitales] [Convocatoria en Word] Caso del Hospital Universitario de Getafe Dra. Laura Nájera
Diagnóstico: LEIOMIOSARCOMA Historia Clínica Mujer de 56 años, sin antecedentes de interés, que acude a nuestro hospital por presentar una masa en hemiabdomen izquierdo de crecimiento progresivo acompañada de naúseas, vómitos y estreñimiento. El estudio radiológico mostró una gran masa con captación irregular y heterogénea de contraste. Se sugirió un sarcoma abdominal con dependencia gástrica. Se realizó extirpación de la lesión. A los 2 años del diagnóstico la enferma presentó LOES hepáticas (diapositiva enviada), que se diagnosticaron como metastáticas.
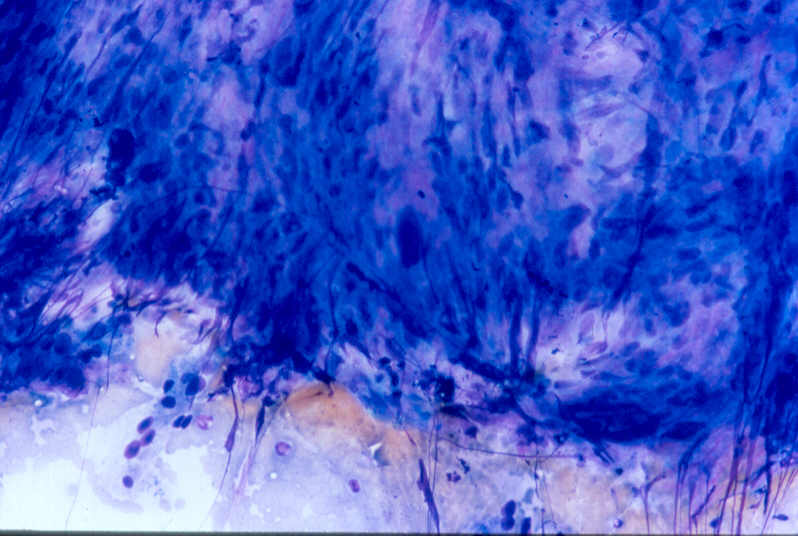
Anatomía Patológica La masa (360 gramos, 11,5 x 9 x 9 cm) era polilobulada y estaba delimitada por una fina cápsula. Al corte, presentaba un tejido sólido, blanquecino y firme, de aspecto trabeculado, con áreas geográficas de aspecto necrótico. Histológicamente la lesión estaba constituida por una proliferación densamente celular de patrón fasciculado, de células con marcado pleomorfismo, con núcleos ovoides con ocasionales nucleolos prominentes. El citoplasma era eosinófilo y mal definido y se entremezclaban focos de necrosis (<15% de la totalidad de la neoplasia). El recuento mitótico fue de 9/10 cga. El estudio inmunohistoquímico mostró positividad intensa para marcadores musculares (desmina, actina de músculo liso y actina de músculo específica). Fueron negativos CD-34, S-100 y c-Kit. Comentario Los leiomiosarcomas son neoplasias mesenquimales malignas con diferenciación de músculo liso. Constituyen entre el 5-10% de los sarcomas de partes blandas. La etiología es desconocida. Se clasifican según la localización debido a diferencias clínicas y biológicas en intraabdominales, subcutáneos, de origen vascular y relacionados con inmunodeprimidos. Dentro de los intraabdominales, los de retroperitoneo son los más frecuentes. Dos terceras partes son mujeres y la edad de presentación es la quinta década de la vida. La clínica es inespecífica, siendo frecuentes el dolor, la presencia de masa, pérdida de peso, naúseas o vómitos. Suelen ser tumores grandes que afectan otros órganos y quirúrgicamente irresecables. Dentro de los criterios de malignidad se encuentran el tamaño de la lesión, la atipia celular, la celularidad y la presencia de necrosis. Sin embargo, el criterio más importante es la actividad mitótica. Se consideran tumores malignos cuando hay 5 mitosis/10 cga, tumores con potencial maligno si hay entre 1-4 mitosis/10 cga y, si el recuento es de 0-5/50 cga, el diagnóstico será de benignidad o de potencial maligno dependiendo de la presencia o no de atipia celular. Los hallazgos ultraestructurales son inespecíficos y hay que correlacionarlos siempre con la histología. En cuanto a la citogenética los cariotipos son complejos y no característicos, con pérdida o ganancia de regiones cromosómicas. La vía de la Rb-ciclina D está alterada en un 50% de los casos y se han encontrado mutaciones de la proteína p53 en un 15-50%; esto último parece asociar un peor pronóstico. El principal diagnóstico diferencial en esta localización hay que realizarlo con los tumores del estroma gastrointestinal, si bien estos son positivos con CD-34 y con c-Kit. El tratamiento de elección es la extirpación quirúrgica y el pronóstico depende fundamentalmente de la localización y el tamaño; tumores mayores de 10 cm y retroperitoneales presentan peor pronóstico. El grado histológico, la edad (peor en > de 60 años) y la resección quirúrgica completa con márgenes libres también influyen en el pronóstico. Recurrencias locales se dan en un 10-25% y las metástasis a distancia en un 45%, siendo los lugares más frecuentes el pulmón y el hígado. La supervivencia global es del 61% a los 5 años del diagnóstico. Bibliografía "Soft Tissue Tumors" Enzinger and Weiss´s. Fourth Edition (págs 728-741) "Tumors of Soft Tissue and Bone". WHO Classification of Tumors (págs. 131-134) "Tumors of the Soft Tissues". Atlas of Tumor Pathology. AFIP (págs. 239-256)
[Convocatoria] [Programa] [Hospitales] [Convocatoria en Word]
|
. | |
| © SEAP. Sociedad Española de Anatomía Patológica | Actualizado: 27/08/2004 |
||