|
Información || Congresos || Cursos || Territoriales || Noticias || Patología || Telepatología |
|
Información || Congresos || Cursos || Territoriales || Noticias || Patología || Telepatología |
| . |
[Noticias]
[Boletín de la SEAP] [Bolsa
de Trabajo]
[Convocatoria] [Programa] [Hospitales] [Convocatoria en Word] HOSPITAL CLÍNICO SAN CARLOS. MADRID. Caso clínico 1. Juan Ruiz, Noemí García, Lucía González, Armando Martínez.
HISTORIA CLÍNICA Niño de 8 años con ptosis palpebral congénita intervenida en 5 ocasiones. Refiere dolor intenso dorsal desde hace tres meses y dificultad en la marcha de tres días de evolución, por lo que ingresa. A la exploración física presenta leve disminución de fuerza con reflejos de estiramiento exaltados, aumento del área reflexógena, clonus y Babinski positivo. Los reflejos superficiales cremastérico y cutáneo abdominal están abolidos. La sensibilidad está disminuida por debajo del abdomen, con nivel sensitivo dorsal D5-D6. Presenta marcha torpe de características paréticas, con signo de segador. RMN de columna: lesión intradural extramedular de 2x1 cm. que ocupa gran parte del canal medular. Desplazamiento postero-lateral izquierdo de la médula a nivel de D4. Realce homogéneo con la administración de contraste. Diagnóstico clínico: meningioma. Pieza quirúrgica: masa en canal medular a nivel de D4, anterolateral derecha, adherida a duramadre, consistencia dura y poco sangrante.
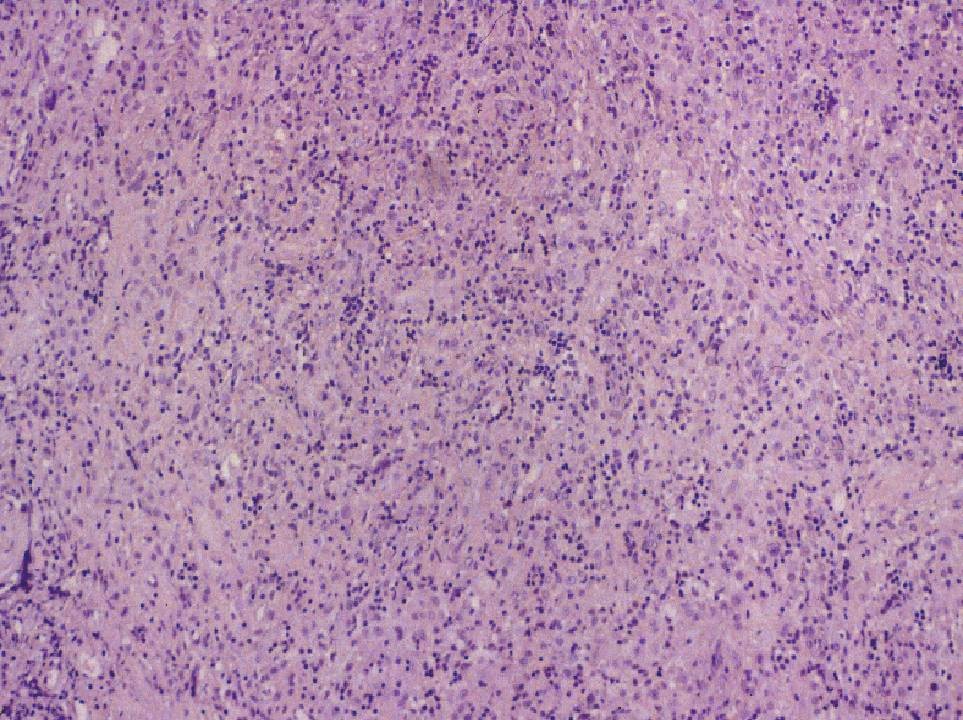
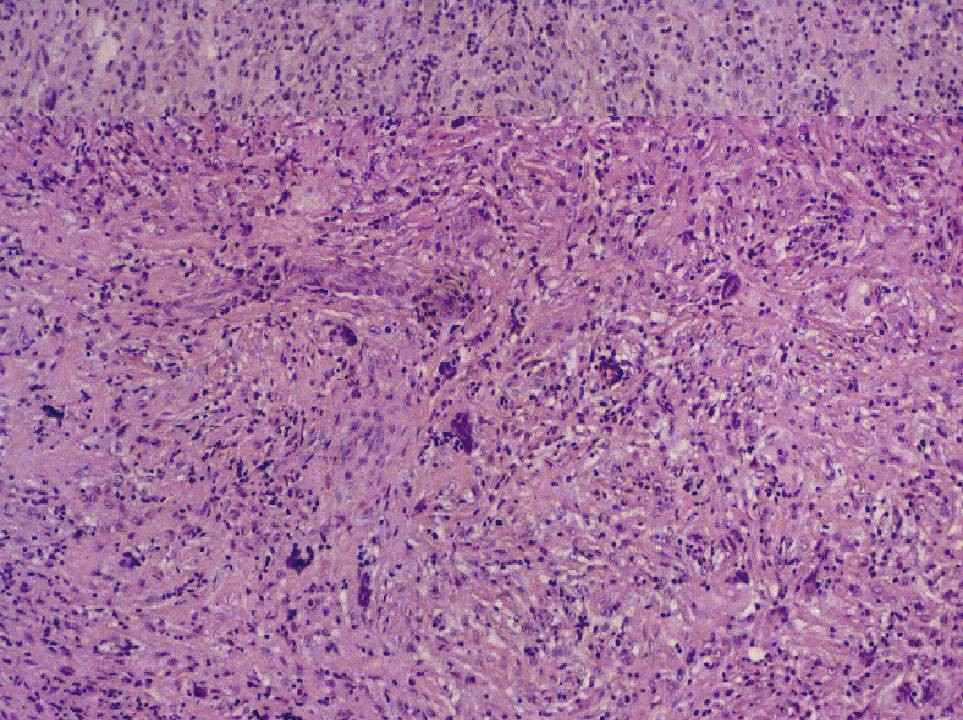
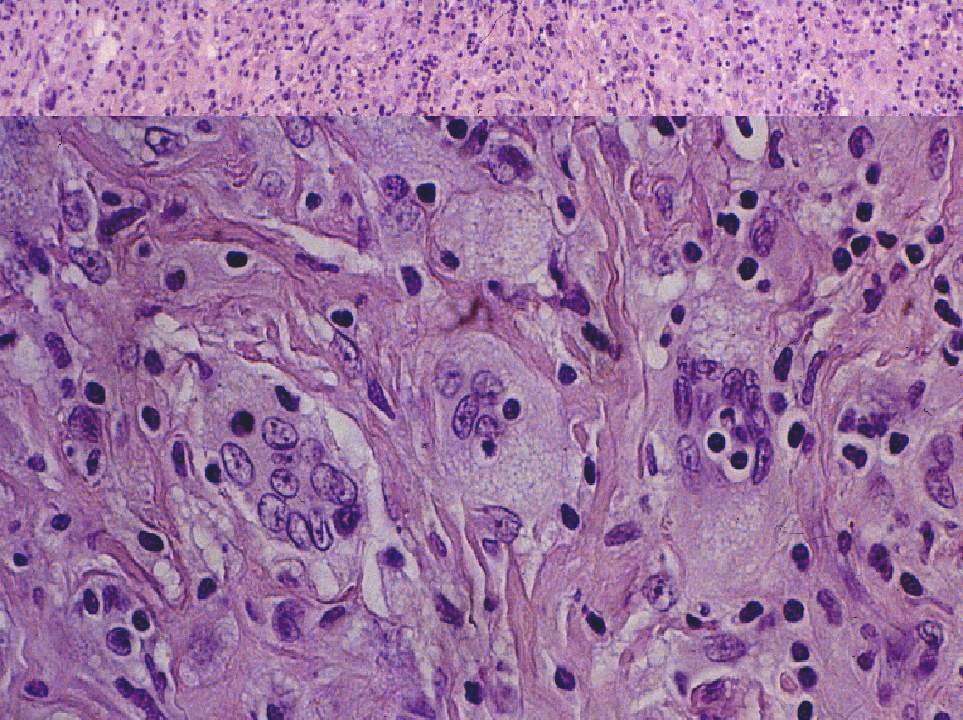
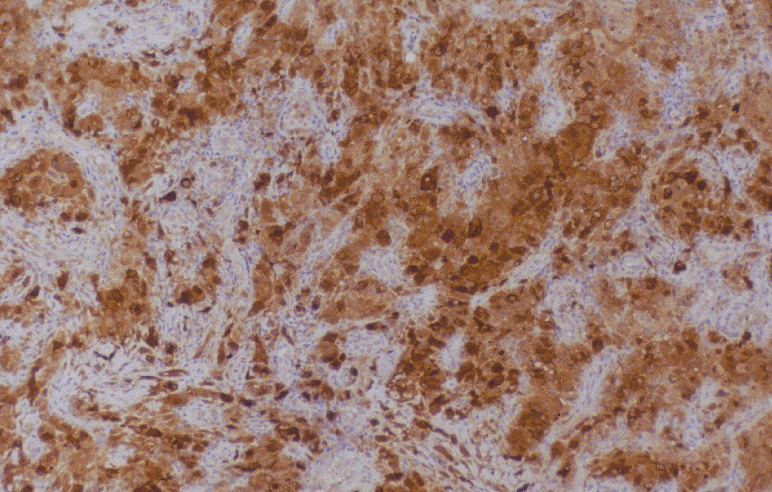
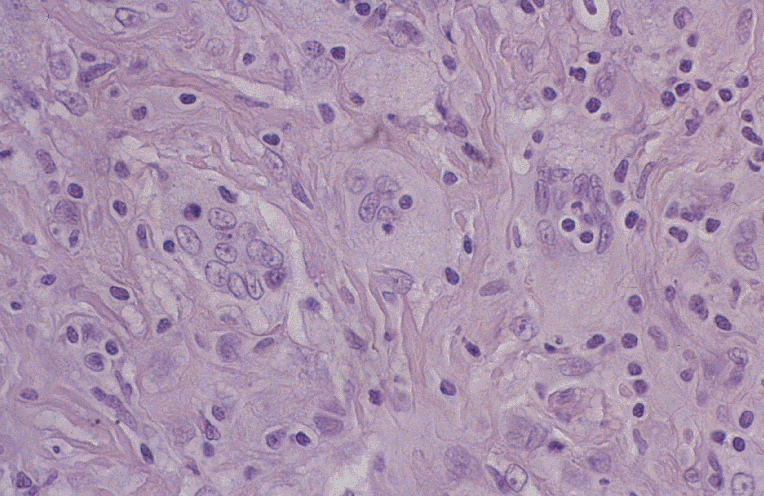
DIAGNÓSTICO ANATOMOPATOLÓGICO Enfermedad de Rosai-Dorfman extranodal. Estudio macroscópico: múltiples pequeños fragmentos de consistencia fibrosa y coloración amarillenta, que en conjunto miden 1.5 cm. de diámetro. Estudio microscópico: se observa una proliferación celular de histiocitos de talla grande, con citoplasma amplio, a veces vacuolado y núcleo vesiculoso. Se observan algunas células multinucleadas. Ocasionalmente hay eritrocitos y linfocitos intracitoplásmicos (emperipolesis). Estas células muestran el siguiente fenotipo con técnicas de inmunohistoquímica: CD68 +, proteína S-100 +, fascina +, CD34 -, CD31 -, CD1a -. El fondo está constituido por linfocitos de talla pequeña de fenotipo B (CD20 +) y T (CD3 +), así como por células plasmáticas inmersas en un estroma fibroso colagenizado.
Fig. 1. Emperipolesis. Fig. 2. Proteína S-100 DISCUSIÓN La enfermedad de Rosai-Dorfman (ERD) fue descrita por primera vez por estos autores en 1969 (1), con el nombre de histiocitosis sinusal con linfadenopatía masiva. Actualmente se clasifica dentro de las histiocitosis de macrófagos reactivas con hemofagocitosis (2, 3). Se trata de una enfermedad idiopática que afecta habitualmente a menores de 20 años, con predominio en raza negra. La forma típica de la enfermedad cursa con linfadenopatía masiva bilateral, no dolorosa, preferentemente en cuello, con fiebre, leucocitosis, aumento de VSG e hipergammaglobulinemia policlonal. Uno de cada cuatro casos tienen afectación extranodal que puede o no acompañar a la linfadenopatía. La afectación extranodal está descrita en órbita, tracto respiratorio superior, cabeza y cuello, piel y SNC, dentro del cual puede localizarse intracranealmente, en leptomeninge, o como en este caso, en duramadre espinal. (4,5).
El diagnóstico diferencial se plantea con diversos procesos. Un dato morfológico de gran ayuda es la presencia de emperipolesis en la ERD. En la histiocitosis sinusal inespecífica, aparte de la estructura general de la lesión, no existe amperipolesis ni inmunoreactividad a la proteína S-100. La histiocitosis de células de Langerhans presenta positividad para CD1a y negatividad para fascina. La histiocitosis sinusal inducida por cobalto-cromo y titanio tras sustitución protésica, es el cuadro que morfológicamente más se asemeja a la ERD, pero además del antecedente de cambio protésico, aparece en los ganglios pélvicos (6). El xantogranuloma se caracteriza por la presencia de histiocitos vacuolados que son negativos para proteína S-100. Debido a su rareza, el comportamiento biológico de esta enfermedad no es bien conocido, sin embargo, la resección total de la lesión parece ser curativa (2), como ocurre en nuestro caso, que no presenta remisión hasta la fecha.
BIBLIOGRAFÍA 1. Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. A newly recognized benign clinicopathological entity. Arch Pathol 1969; 87: 6370. 2. Foucar E, Rosai J, Dorfman RD. Sinus histiocytosis with massive lymphadenopaty (Rosai-Dorfman Disease): Review of the entity. Semin Diagn Pathol 1990; 7:19-73. 3. Woda BA, Sullivan JL. Reactive histiocytic disorder. Am J Clin Pathol 1993; 99:459-463. 4. Andriko JA, Morrison JA, Colegial CH, Davis BJ, Jones RV. Rosai-Dorfman disease isolated to the central nervous system: a report of 11 cases. Mod Pathol 2001;14:172-178. 5. Foucar E, Rosai J, Dorfman RF, Brynes RK. The neurologic manifestations of sinus histiocytosis with massive lymphadenopathy. Neurology 1982; 32:365-371. 6. Albores-Saavedra J, Vuitch F, Delgado R, Wiley E, Hagler H. Sinus histiocitosis of pelvic lymph nodes after hip replacement. A histiocytic proliferation induced by cobalt-chromium and titanium. Am J Surg Pathol 1994;18:83-90.
[Convocatoria] [Programa] [Hospitales] [Convocatoria en Word]
|
. | |
| © SEAP. Sociedad Española de Anatomía Patológica | Actualizado: 27/08/2004 |
||