|
Información || Congresos || Cursos || Territoriales || Noticias || Patología || Telepatología |
|
Información || Congresos || Cursos || Territoriales || Noticias || Patología || Telepatología |
| . |
[XXII Congreso Nacional (Palma de Mallorca)] [XXI Congreso Nacional (Madrid)] [ XX Congreso Nacional (Pamplona) ] [XIX Congreso Nacional (Barcelona) ] [XVIII Congreso Nacional (Málaga) ]
SEMINARIO DE PATOLOGÍA ÓSEA
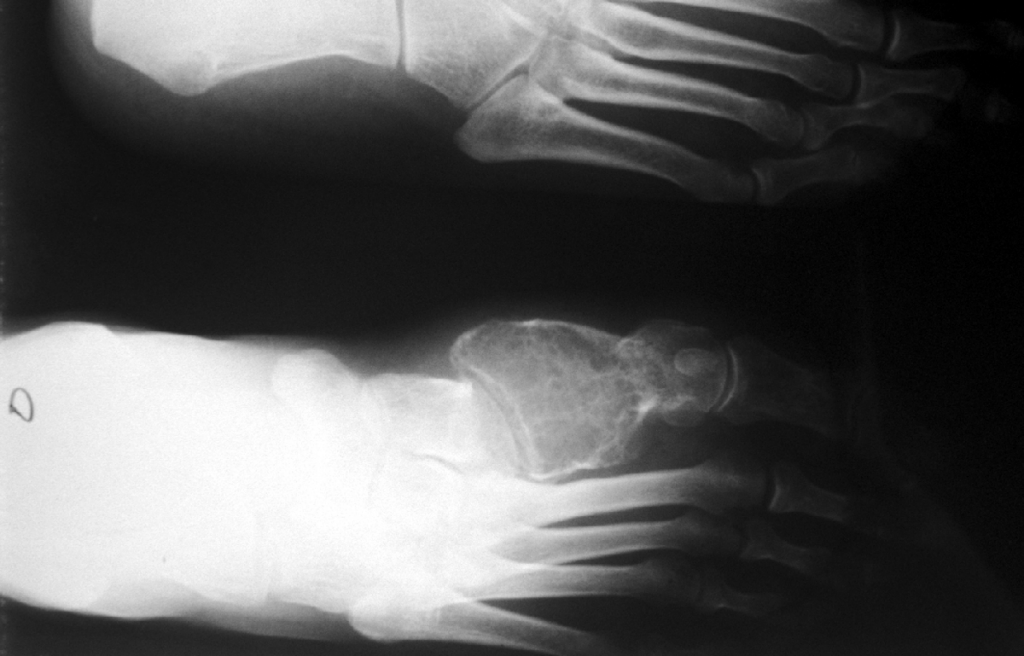
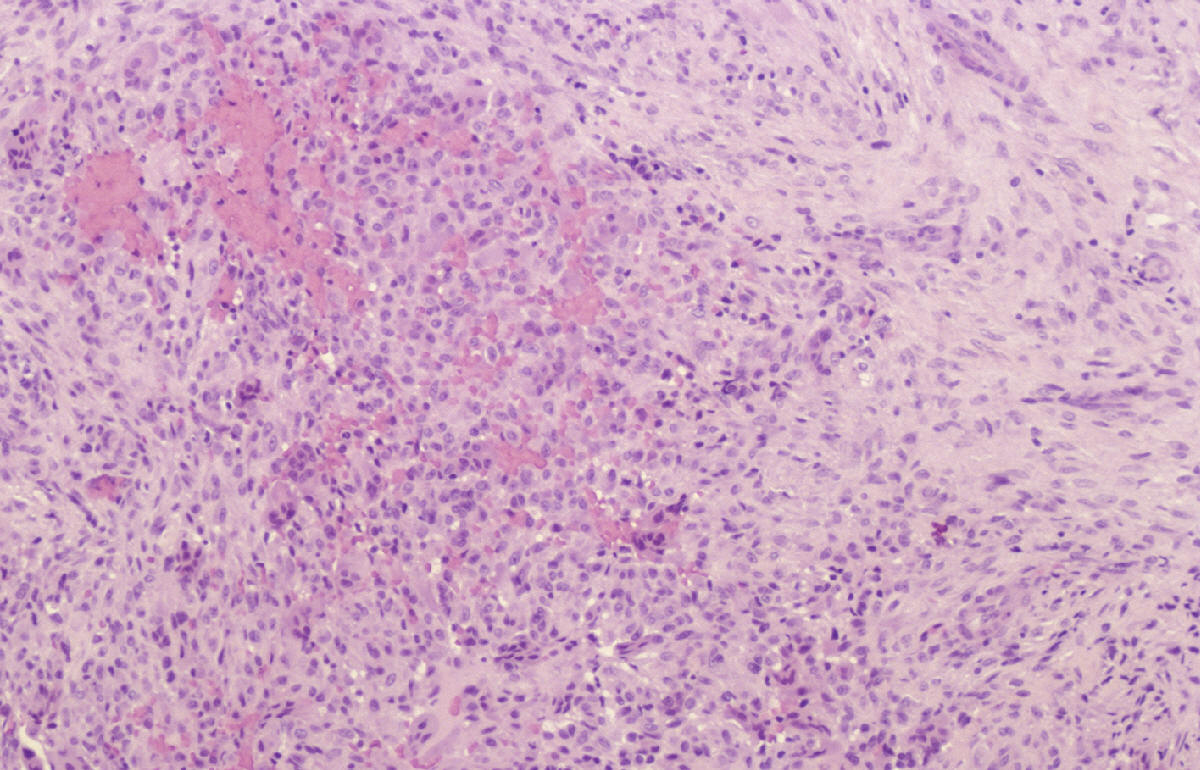
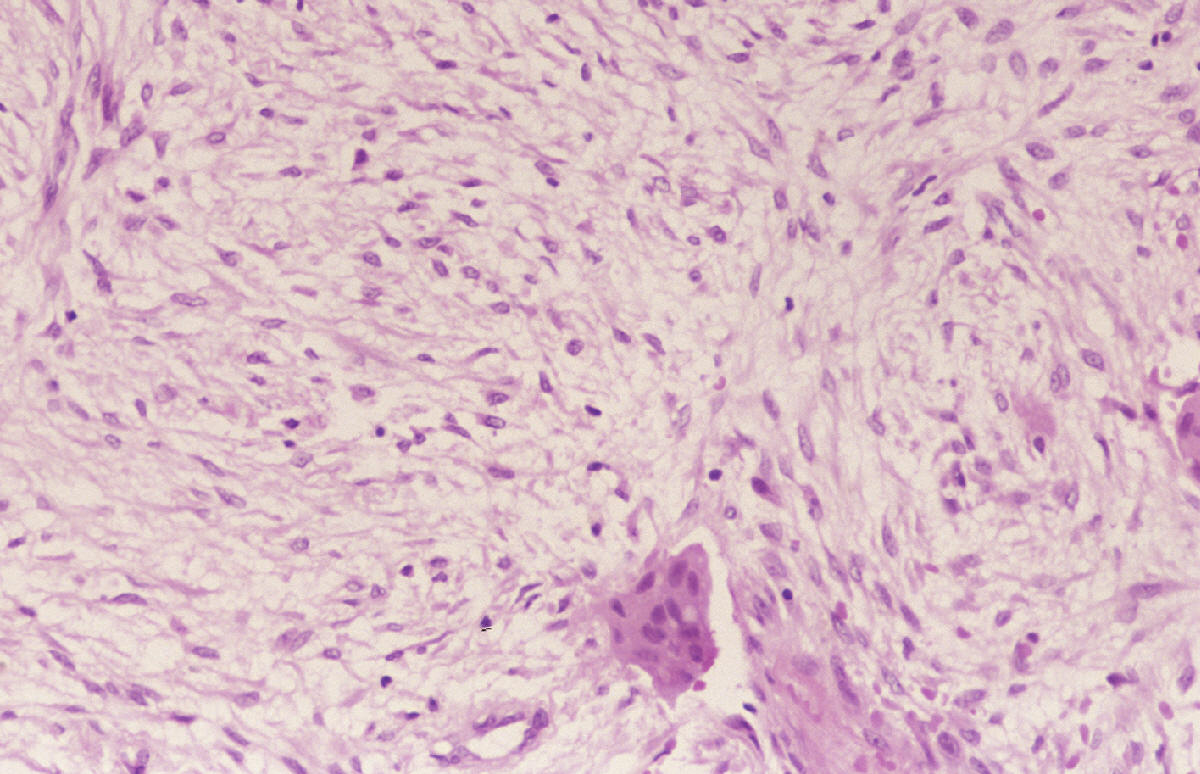
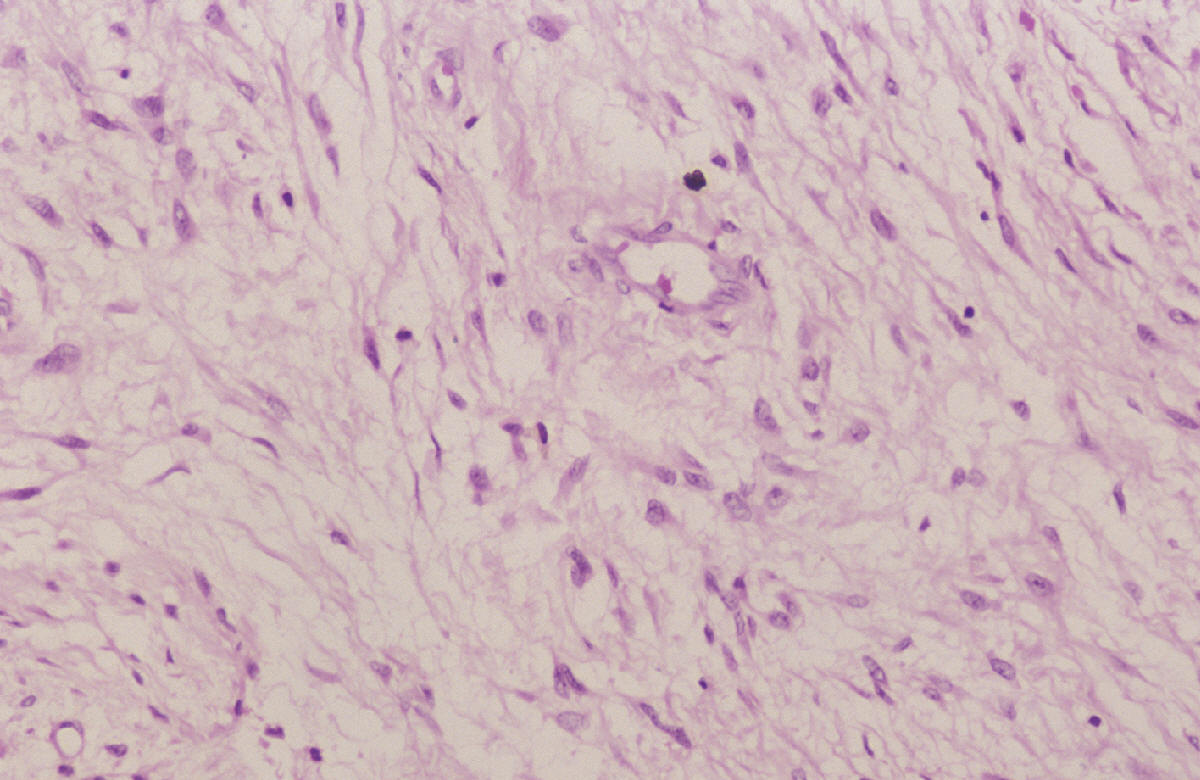
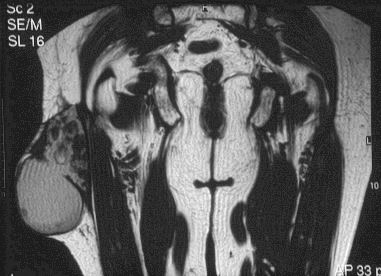
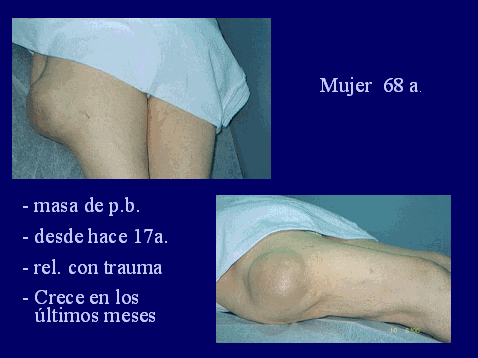
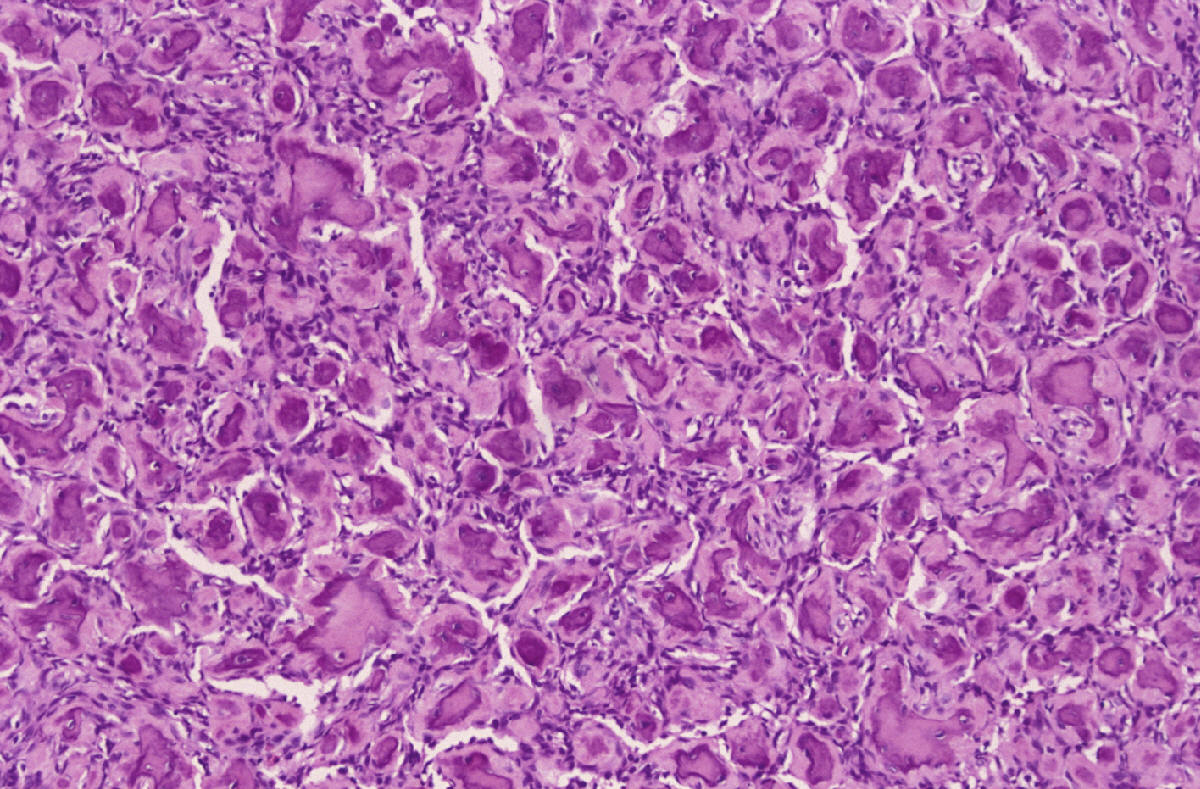
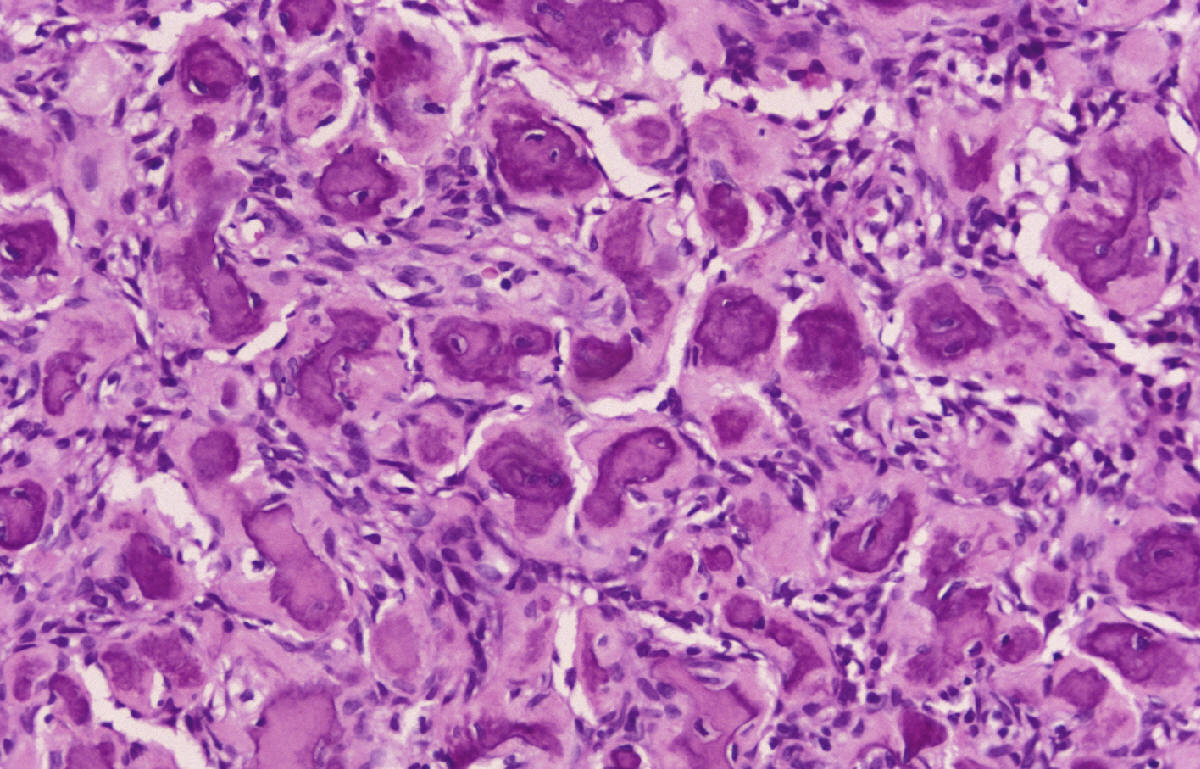
SALA MUNICH Dr. F.J. Martínez Tello SEMINARIO. HISTORIAS CLÍNICAS CASO 1 Dr. J.M. Loizaga Iriondo CASO 2 Dr. J. Pérez Villanueva CASO 4 Dr. J.C. Lorenzo Roldán CASO 6 Dra. I. González Mediero CASO 8 Dr. F.J. Martinez Tello CASO Nº 1.- Dr.J.M.Loizaga Iriondo CASO Nº 2.- Dr.J.Pérez Villanueva CASO Nº 3.- Dr.M.Calvo Asensio En el examen físico se aprecia un tumor de 10 cm de diámetro, localizado en el tercion superior de húmero izquierdo, duro y no adherido a piel. No adenopatias pero si dolor a la presión. CASO Nº 4.-Dr.J.C.Lorenzo Roldán Antecedentes: traumatismo en esta región hace 17 años, con la aparición de un "bulto", indoloro, de 4 cm. de diámetro que ha permanecido estable hasta que hace 6 meses comenzó a crecer. A la exploración: Tumoración de aproximadamente 14 cm. de eje mayor, indolora. Radiología simple y RMN. Las preparaciones histológicas (2) corresponden a la pieza de exéresis de la tumoración. CASO Nº 5.- Dr.F.López Barea CASO Nº 6.- Dra.I.Gonzalez Mediero Los síntomas han ido aumentando con incurvación anterior de la tibia, observándose radiológicamente una extensa lesión diafisaria anterior lítica y multifocal con bordes esclerosos y predominantemente intracortical. La lesión es resecada y curetada en su base, rellenándose y fijándose con Ortofix. CASO Nº 7.- Dr. J.Vila Torres CASO Nº 8 .-Dr. F.J.Martinez Tello SEMINARIO: CASO Nº1 .-Dr.J.M.Loizaga Iriondo Proceso reparativo de células gigantes en huesos de manos y pies SINONIMIA: reacción de células gigantes del hueso o granuloma de células gigantes o granuloma reparativo de células gigantes. DEFINICIÓN: Proliferación benigna, probablemente reactiva, caracterizada por la proliferación de células gigantes, típicamente en agregados focales, sobre un fondo de proliferación fibroblástica. Fue descrita por Jaffe en 1958 en los huesos maxilares y por Ackerman y Spjut en 1962 en huesos extramaxilares, concretamente en falanges de las manos y los pies, bajo la denominación de "reacción de células gigantes". Posteriormente se describió también en huesos largos preferentemente en fémur.En 1983 Sanerkin describió con el nombre de forma sólida de quiste óseo aneurismático un proceso semejante que actualmente se considera lo mismo que el proceso reparativo de células gigantes. RADIOLOGICAMENTE se manifiesta como una zona de osteolisis con expansión ósea y ausencia de calcificaciones o de trabeculación intralesional. LA HISTOLOGIA se caracteriza por una proliferación fibroblastica en un estroma fibroso y por acúmulos de osteoclastos en agrupaciones preferentemente alrededor de áreas hemorrágicas y típicamente en la periferia de las lesiones. En ocasiones se asocian a quistes óseos aneurismáticos con grandes dilataciones vasculares, pared fibrosa y células gigantes. Pueden ser múltiples en cuyo caso el diagnóstico diferencial se plantea con el hiperparatiroidismo, y con lesiones semejantes con proliferación de osteoclastos y estroma fibroso que pueden ocurrir en la enfermedad de Paget. La caracterización fenotípica de las células gigantes con CD86 positivo, fosfatasa ácida tartrato resistente positiva CD45 y positividad a vitronectina, identifican estas células con los osteoclastos. Las células mononucleadas se comportan en cultivo de tejidos como positivas para la fosfatasa alcalina y expresan el activador-receptor NFG ligando kappa B (RANKL). Por todo ello se interprata que los osteoclastos estan formados por células del sistema monocítico/macrofágico precursoras de osteoclastos que se diferencian en tales por la acción de células mononucleares de fenotipo osteoblástico asentadas en el estroma de estos tumores. BIBLIOGRAFIA: 1.-Jaffe HL. Tumors and tumorous conditions of the bones and joints.1958. A textbook. Lea and Febiger. Philadelphia. Pp 49-50 2.-Ackerman LV y Spjut HJ. Tumors of bone and cartilage. Atlas of Tumor Pathology, section II-fascicle IV. Washington DC. Armed Forces Institute of Pathology. 1962: 344-345. 3.-Lorenzo JC y Dorfman HD. Giant cell reparative granuloma of shorf tubular bones of the nads and feet. Am J. Surg.Pathol. 1980; 4:551-563. 4.-Ratner V y Ddorfman HD. Giant cell reparative granuloma of the hand and feet. Clin. Orthip. 1990; 260: 251-258. 5.-Yamaguchi T y Dorfman HD.Int.J.Surg. Pathol. 2001 9(3):189-200 6.-Itonaga I, Shulze E y col. Phenotypic characterization of mononuclear and multinucleated cells of giant cell reparative granuloma of small bones. J. Pathol. 2002; 198(1):30-36. 7.-Caskey PM, Wolf MD y Fechner RE. Multicentrix giant cell reparative granuloma of small bones of the hand. Clin. Orthop.1985; 193: 199-205. 8.-Sanerkin NG, Morr MG y Roylance MB. An unusual intraosseous lesion with fibrobastic, osteoblastic, Aneurysmal y fibromixoid elements. Cancer 1983, 51:2278-2286.
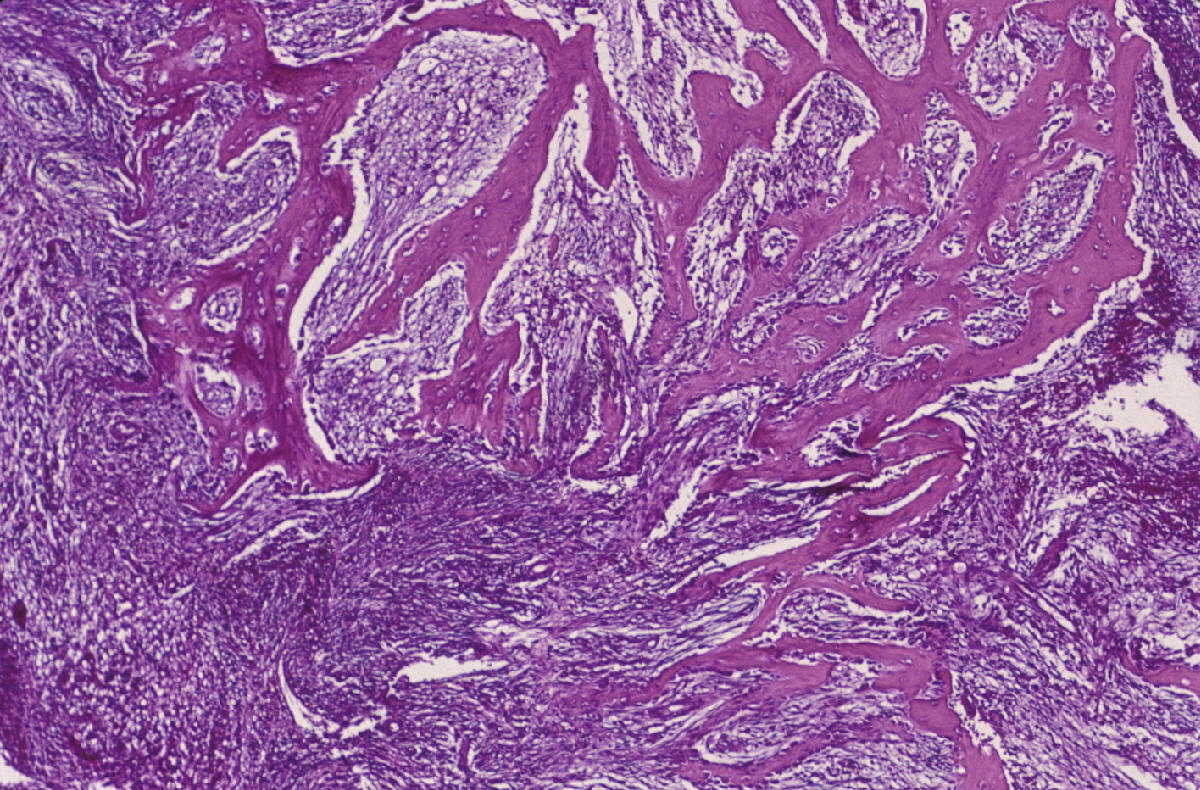
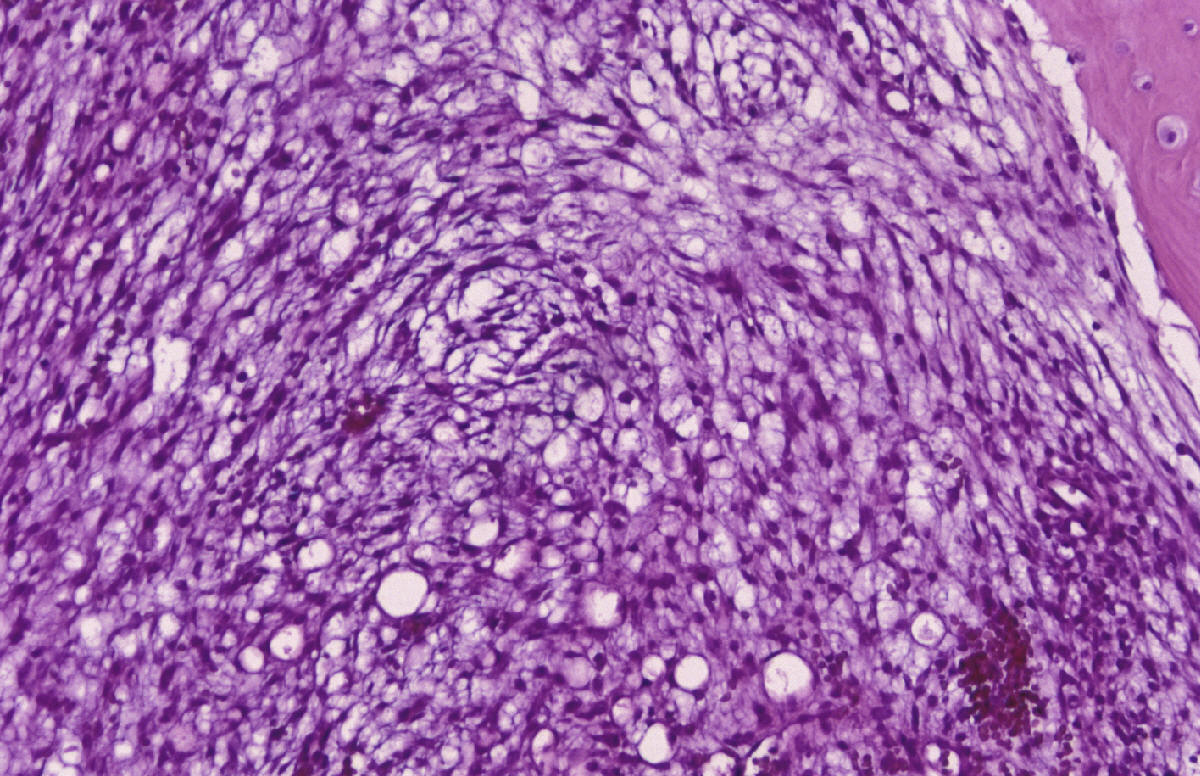
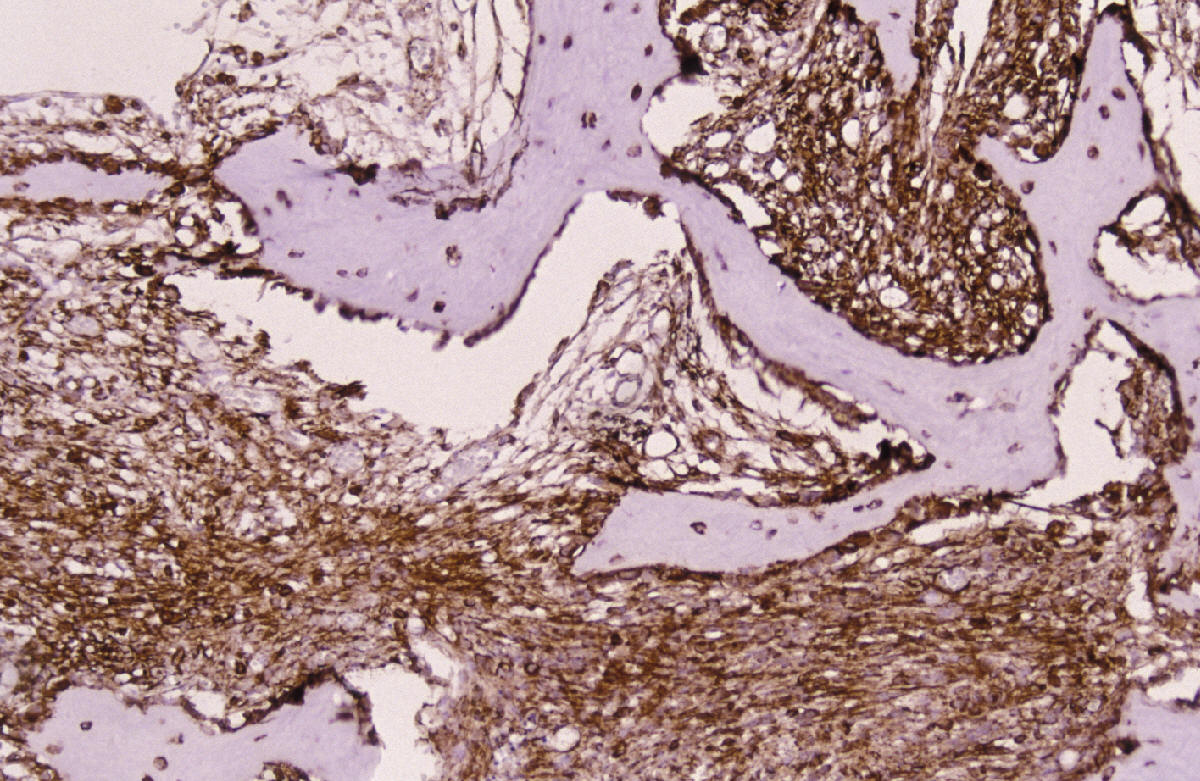
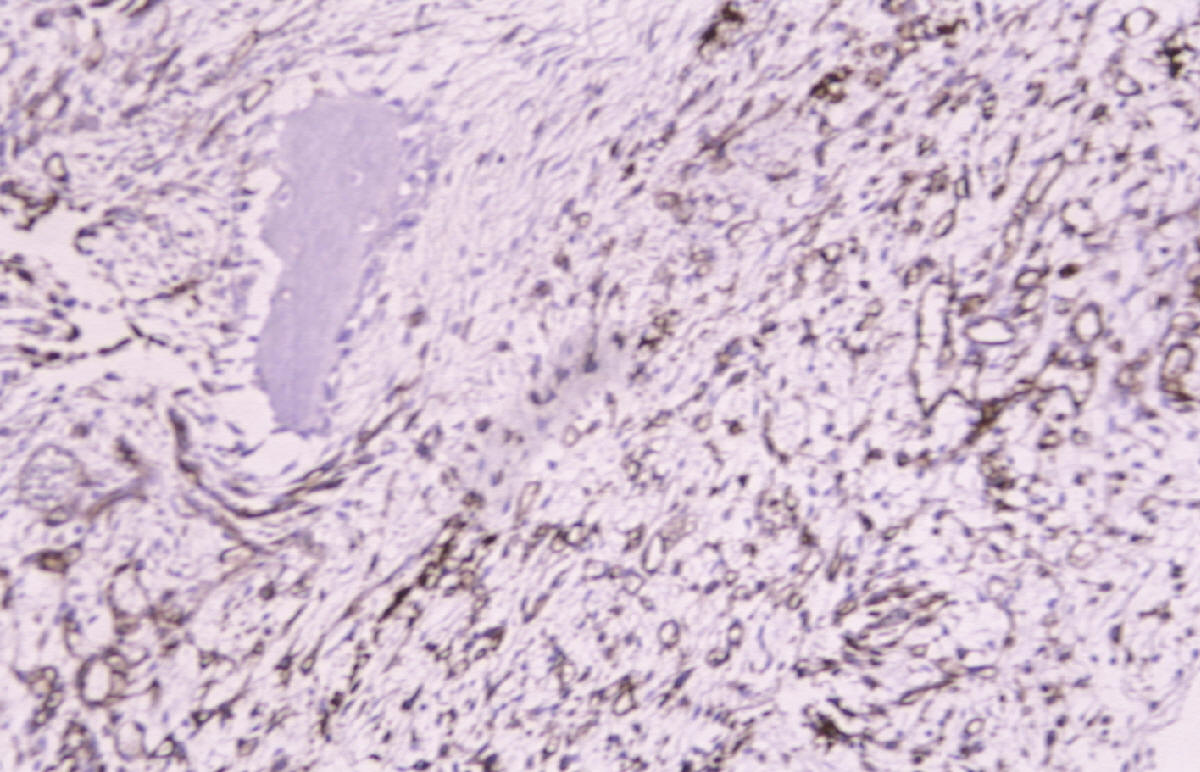
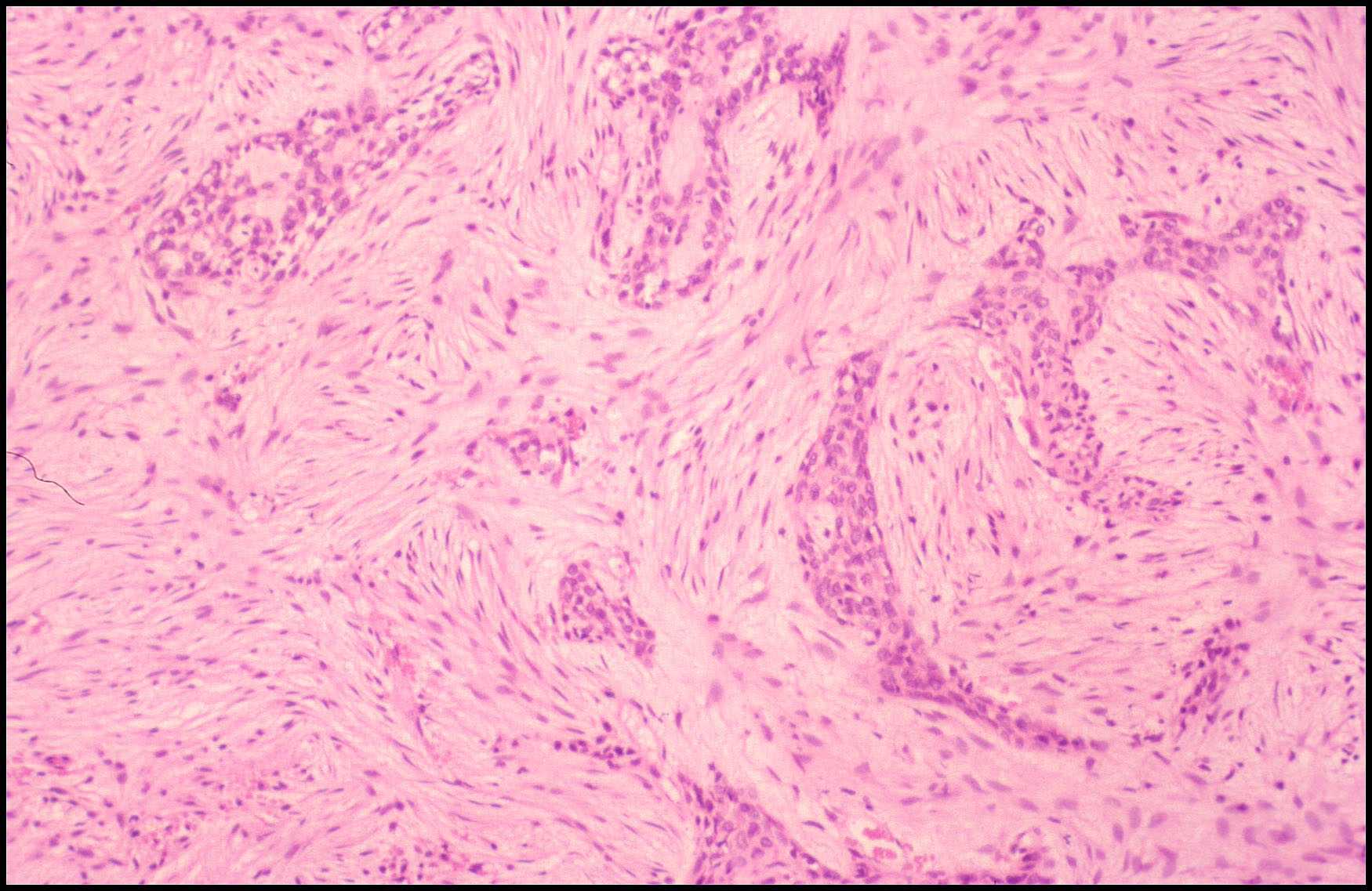
CASO Nº 2 Dr.J.Pérez Villanueva. Historia Clinica: Varón de 32 años con síntomas radiculares en región torácica dorsal y ligera escoliosis. Técnicas de exploración por imagen evidenciaron una lesión afectando región posterior de cuerpo de la octava vertebra torácica así como parte adyacente del pediculo dercho, ampliación del espacio epidural contiguo y discreta latreralización y compresión medular. Se realizó intervención qurúrgica resecándose tumor de 2,5 cm. que afectaba la parte posterior de cuerpo vertebral y pediculo adyacente así como espacio epidural. Microscópicamente la lesión presentaba de una parte un componente óseo muy llamativo en forma de trabéculas ricamente anatomicas, frecuentemente con ribete osteoblástico variablemente prominente, en ocasiones epitelioide, y de otra parte componente tisular blando intertrabecular en forma de patrón mixto incluyendo tejido conectivo laxo y edematoso con numerosas luces vasculares con endotelios bajos no atípicos, algún espacio vascular más dilatado, y en conjunto el patrón de tejido conectivo laxo intensamente vascularizado compatible con lo esperable en el componente blando intertrabecular de un osteoblastoma. Sin embargo, otras zonas mostraban un patrón más sólido, o al menos mixto, con áreas de patrón más densamente celular a base de elementos de hábito fusiforme con núcleos en general romos presencia en algunas de las células de una pequeña vacuola. En algunos campos se abandonaba el patrón "osteoblastoma-like", al carecer de componente óseo y disponerse en extensiones de proliferación fusocelular sólida con ocasionales luces capilares o hendiduras vasculares intercaladas, algún depósito de hemosiderina y, en conjunto un patrón "kaposiforme". En resumen nos encontramos pues con un cuadro lesional que en unos sectores presentaba un patrón fuertemente reminiscente de un verdadero osteoblastoma, mientras que en otros la parte no ósea departia notablemente del espectro permisible en esa entidad, especialmente las zonas sólidas fusocelulares. Estos sectores blandos no compatibles con tal entidad ósea primaria nos hicieron considerar como diagnósticos diferenciales entidades tales como cordoma de variante fusocelular; meningioma cordoide y la entidad originalmente denominada hemangioendotelioma fusocelular y, más recientemente, rebautizada como hemangioma fusocelular. Las áreas claramente vasculares, o de patrón mixto luminizado/sólido, ayudaron considerablemente a decantarnos desde el punto de vista puramente morfopatológico por un hemangioma fusocelular. En el perfil inmunohistoquímico destacaron la fuerte positividad para vimentina y para CD 31, siendo negativos los marcadores que hubiesen apoyado las otras entidades propuestas. Nos encontramos pues ante un caso de patologia ósea doblemente interesante. Son muy pocos los casos de hemangioma fusocelular primario de hueso descritos hasta la fecha, pero el interés del caso se acrecienta notablemente por la espectacular reacción osteoformativa mimetizante en muchos campos de un osteoblastoma ("osteoblastoma-like reaction"). Esta reacción en forma de pseudoosteoblastoma ha sido descrita como respuesta ósea del huésped a otras entidades nosológicas pero no ante un hemangioma fusocelular; dicha reaccción forma parte del capítulo de pseudotumores óseos siendo en ocasiones tan florida que puede, si no se analizan criticamente las áreas no óses de la lesión, precipitar un falso diagnóstico de verdadero osteoblastoma. La reacción de tipo pseudo-osteoblastoma, se genera preferentemente como respuesta del hueso huésped a otra lesión, generalmente pero no en exclusiva de tipo tumoral que constituye la lesión primaria. Alternativamente y, con mucha menos frecuencia, la reacción pseudo-osteoblastomatosa puede responder a un fenómeno metaplasico suscitado "de novo en el seno de la propia lesión primaria incitante. Esta segunda modalidad se ha descrito por ejemplo en variantes de meningiomas metaplásicos. El hemangioma fusocelular fue descrito en 1986 por Weis y Enzinger como hemangioendotelioma fusocelular ocurriendo preferentemente en los tejidos superficiales (dermis y subcutis) de las porciones distales de las extremidades, especialmente en la mano. Son muy escasos los ejemplos primarios de hueso descritos. Originalmente fueron considerados como tumores malignos de bajo grado. En las series más recientes los datos de seguimiento aportado indican sin embargo una conducta sistemáticamente benigna tras la excisión adecuada. El potencial biológico exacto en el caso especifico de asiento en hueso no está totalmente determinado. No hay predilección de sexo pudiéndose presentar en un amplio espectro de edad con tendencia a concentración en las décadas segunda y tercera. El curso clínico es indolente y las lesiones nueves que originalmente fueron interpretadas como recurrencias, parecen corrresponder a lesiones que surgen de novo. Infrecuentemente se ha evidenciado regresión espontánea. Pueden asociarse con síndrome de Mafucci's o síndrome de Klippel-Trenaunay. Algunos autores consideran que el hemangioendotelioma fusocelular puede no corresponder a una verdadera neoplasia sino una lesión reactiva asociada a anomalías en el flujo sanguíneo local o una malformación vascular. DIAGNOSTICO: Hemangioma fusocelular óseo con reacción tipo pseudo-osteoblastoma. REFERENCIAS: 1.- Weis S W, Enzinger F M 1986 Spindle cell hemangioendothelioma a low grade angiosarcoma resembling a cavernous hemangioma anf Kaposi's sarcoma Am J Surg Pathol 10:521-530- 2.-Dorfman H, Czeniak B: Bone Tumors; Mosby 1998, pag 783-795 3.-Perkins P, Weis S W 1996 Spindle cell hemanigoendothelioma: an analysis of 78 cases with reassessment of its pathogenesis and biologic behavior. Am J Surg Pathol 20: 1196-1204. 4.- Fanburg J C, Meis-Kindblom J M; Rosenberg A C 1995 Multiple enchondromas associated with spindle-cell hemangioendotheliomas. An overlooked variant of Mafucci's syndrome. Am J Surg Pathol 97: 279-287
Caso Nº 3.- Dr. Miguel Calvo Asensio DISCUSION (Hematoma perióstico vs. Periostitis osificante) Se trata de una lesión benigna de características fibro-ósea. No es un verdadero tumor, debiendose considerar una lesión pseudotumoral,dadas las características que posee y que veremos a continuación. Desde el punto de vista clínico observamos que se trata de un paciente con un cuadro de dolor continuo de dos meses de duración, mas acentuado en las dos últimas semanas, en brazo izquierdo, sin astenia, anorexia ni pérdida de peso.La tumoración es grande (10 cm., de diámetro), no se encuentra adherida a la piel, no hay adenopatías axilares y es mas dolorosa a la presión. Radiologicamente se observa un crecimiento de la lesión a partir de la cortical externa del húmero, con escasa participación ó erosión de ésta, con una buena delimitación en los tejidos blandos adyacentes. La lesión afecta a mas de la mitad de la circunferencia del húmero en su periferia. Con éstos datos clínico-radiológicos se podrian barajar una serie de posibilidades diagnósticas, como: 1.Osteosarcoma perióstico. 2. Osteosarcoma parostal. 3.Condrosarcoma perióstico con osificación endocondral benigna. 4. Osteosarcoma de alto grado de la superficie de los huesos. 5.Fibrosarcoma parostal. 6.Osteosarcoma perióstico. Con todos éstos datos se practica una biopsia, que es informada de lesión vascular sin signos de malignidad y posteriormente se efectúa una resección en bloque de toda la lesión. Hay una serie de lesiones benignas óseas ó tumores fibrosos en las que habría que pensar, como son, las periostitis osificantes de huesos largos y planos, de manos y pies (periostitis reactiva florida), la proliferación osteocondromatosa parostal bizarra de manos y pies, la fascitis parostal (fascitis craneal de la infancia y de huesos no craneales), la periostitis dolorosa idiopática, el osteoma osteoide multifocal, perióstico y el osteoblastoma perióstico. Si analizamos las características histológicas de la lesión.vemos que ésta pasa por una serie de fases equivalentes a las del callo de fractura y a las de la miositis osificante. La PERIOSTITIS OSIFICANTE es una de las complicaciones de sangrado en el periostio y ha sido descrita por los diferentes autores con diversos nombres. Entre éstos sinónimos quisiera destacar los mas habituales que encontramos en la literatura, que son: 1.Ligamentotendinitis osificante por avulsión 2.Periostitis post-traumática 3.Hematoma osificante post traumático 4.Miositis osificante post-traumática del periostio 5.Desmoide perióstico 6.Periostitis reactiva florida (Spjut y Dorfman) BIBLIOGRAFIA : -Pathology & Genetics. Tumours of Soft Tissue and Bone. W.H.O. Classifications of Tumours. IARC Press. Lyon, 2002. -Pathology of Bone and Joint Neoplasm. Timothy R.Helliwell, Volume 37 in the Series Major Problems in Pathology.W.B. Sounders Company. 1999. -Colour Atlas of Bone, Joint and Soft Tissue Pathology. Nicholas A. Athanasou. Oxford University Press. 1999.
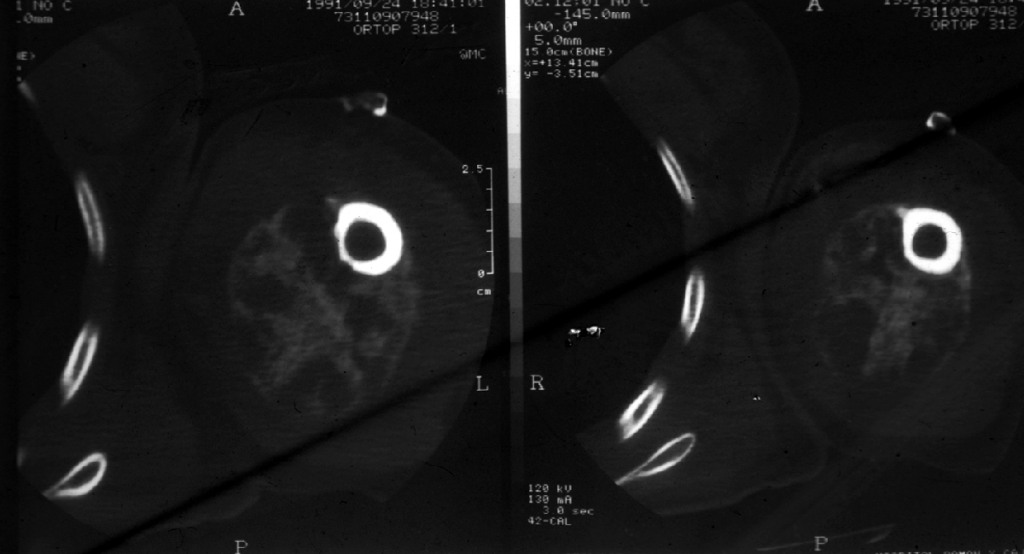
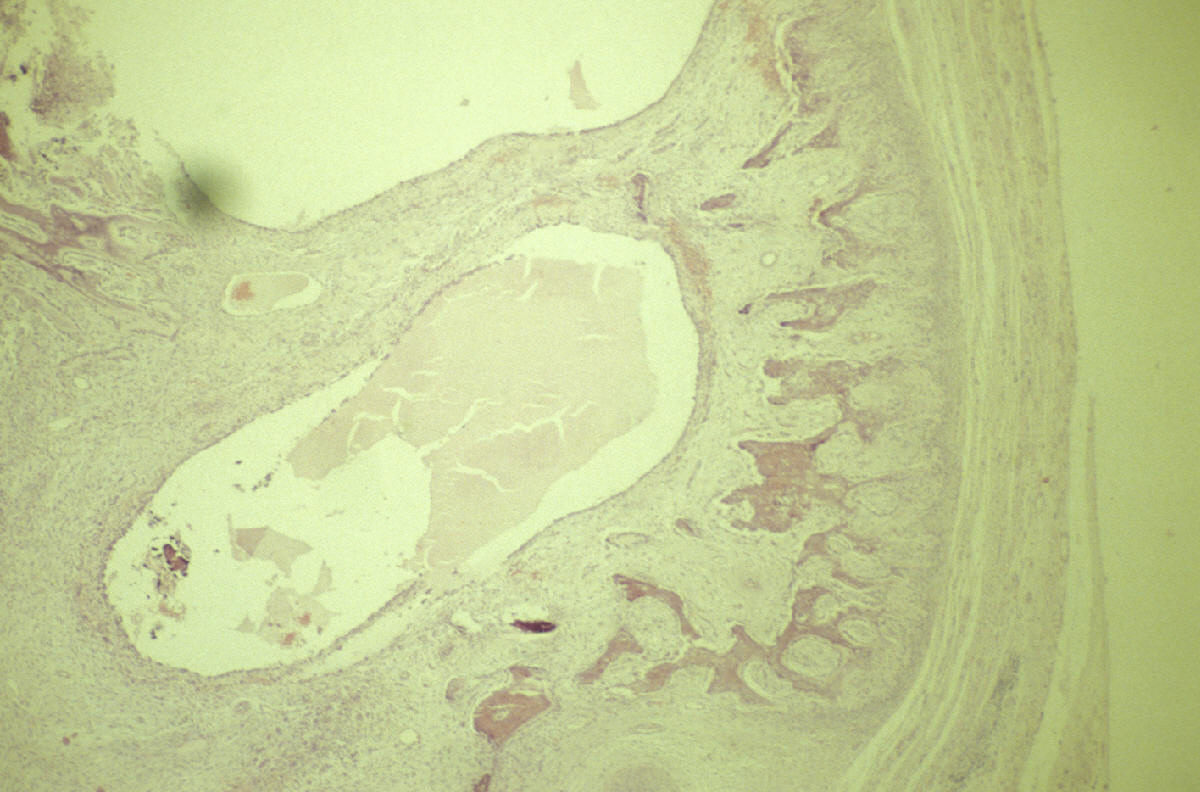
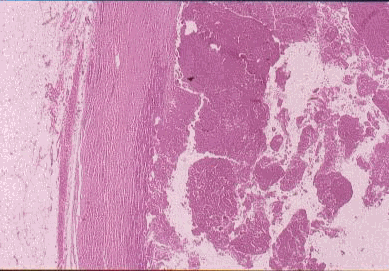
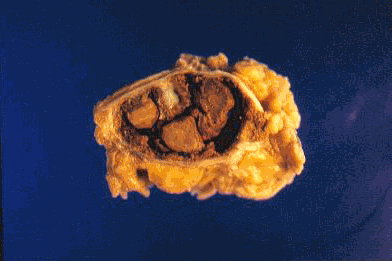
CASO Nº4.-Dr.Juan Carlos Lorenzo Roldan.- Hematoma crónico expansivo Esta lesión que clínica y radiologicamente puede ser confundida con una neoplasia maligna de partes blandas, fue descrita por primera vez como entidad clínico- patológica con este término, en 1980 por REYD et.al.. Posiblemente los casos descritos anteriormente en la literatura como: "Hematoma antiguo" y "Quiste post-traumático de partes blandas" corresponden a la misma entidad. Se trata de un hematoma con un largo período de persistencia, que finalmente aumenta de tamaño progresivamente, creando la sospecha clínica de malignización de un proceso antiguo. Suele haber una historia de trauma o cirugía previa que produjo el hematoma inicial, y se supone que la persistencia y posterior expansión de la lesión es debida a los efectos irritantes del coagulo sanguíneo y a los productos resultantes del proceso de reparación del mismo, que inducirían exudación y sangrado de los capilares del tejido de granulación. Radiologicamente se observa una masa de partes blandas que puede erosionar el hueso adyacente. La Tomografía Axial Computerizada muestra asimismo una masa de partes blandas con ocasional zona calcificada. La RMN muestra una lesión con una zona periférica de baja intensidad de captación y una zona central de alta intensidad de captación. Todos estos cambios no descartan un proceso maligno. Macroscópicamente la lesión está formada por un quiste, generalmente de más de 10 cm. de diámetro, de paredes gruesas y fibrosas y de contenido hemático. Histológicamente no hay sospecha de malignidad. La cápsula del quiste está constituida por tejido colágeno denso con ocasional calcificación distrófica. La luz está ocupada por sangre, fibrina, tejido de granulación con inflamación crónica y calcificación metaplásica. Bibliografía Friedlander HL, Bump RG. "Chronic expanding hematoma of the calf. A case report." J Bone Joint Surg Am. 1968 Sep; 50(6):1237-41. Reid JD, Kommareddi S, Lankerani M, Park MC. "Chronic expanding hematomas. A clinicopathologic entity." JAMA. 1980 Nov 28; 244(21):2441-2. Lewis VL Jr, Johnson PE. "Chronic expanding hematoma." Plast Reconstr Surg. 1987 Mar; 79(3):465-7. Mentzel T, Goodlad JR, Smith MA, Fletcher CD. "Ancient hematoma: a unifying concept for a post-traumatic lesion mimicking an aggressive soft tissue neoplasm." Mod Pathol. 1997 Apr; 10(4):334-40. Aoki T, Nakata H, Watanabe H, Maeda H, Toyonaga T, Hashimoto H, Nakamura T. "The radiological findings in chronic expanding hematoma." Skeletal Radiol. 1999 Jul; 28(7):396-401. Akata S, Ohkubo Y, Jinho P, Saito K, Yamagishi T, Yoshimura M, Kotake F, Kakizaki D, Abe K. "MR features of a case of chronic expanding hematoma." Clin Imaging. 2000 Jan-Feb; 24(1):44-6. Uramoto H, Nakanishi R, Eifuku R, Muranaka H, Takenoyama M, Yoshino I, Osaki T, Yasumoto K. "Chronic expanding hematoma in the chest." J Cardiovasc Surg (Torino). 2000 Feb; 41(1):143-6. Okada K, Sugiyama T, Kato H, Tani T. "Chronic expanding hematoma mimicking soft tissue neoplasm." J Clin Oncol. 2001 Jun 1; 19(11):2971-2. Nakano M, Kondoh T, Igarashi J, Kadowaki A, Arai E. "A case of chronic expanding hematoma in the tensor fascia lata." Dermatol Online J. 2001 Dec; 7(2):6.
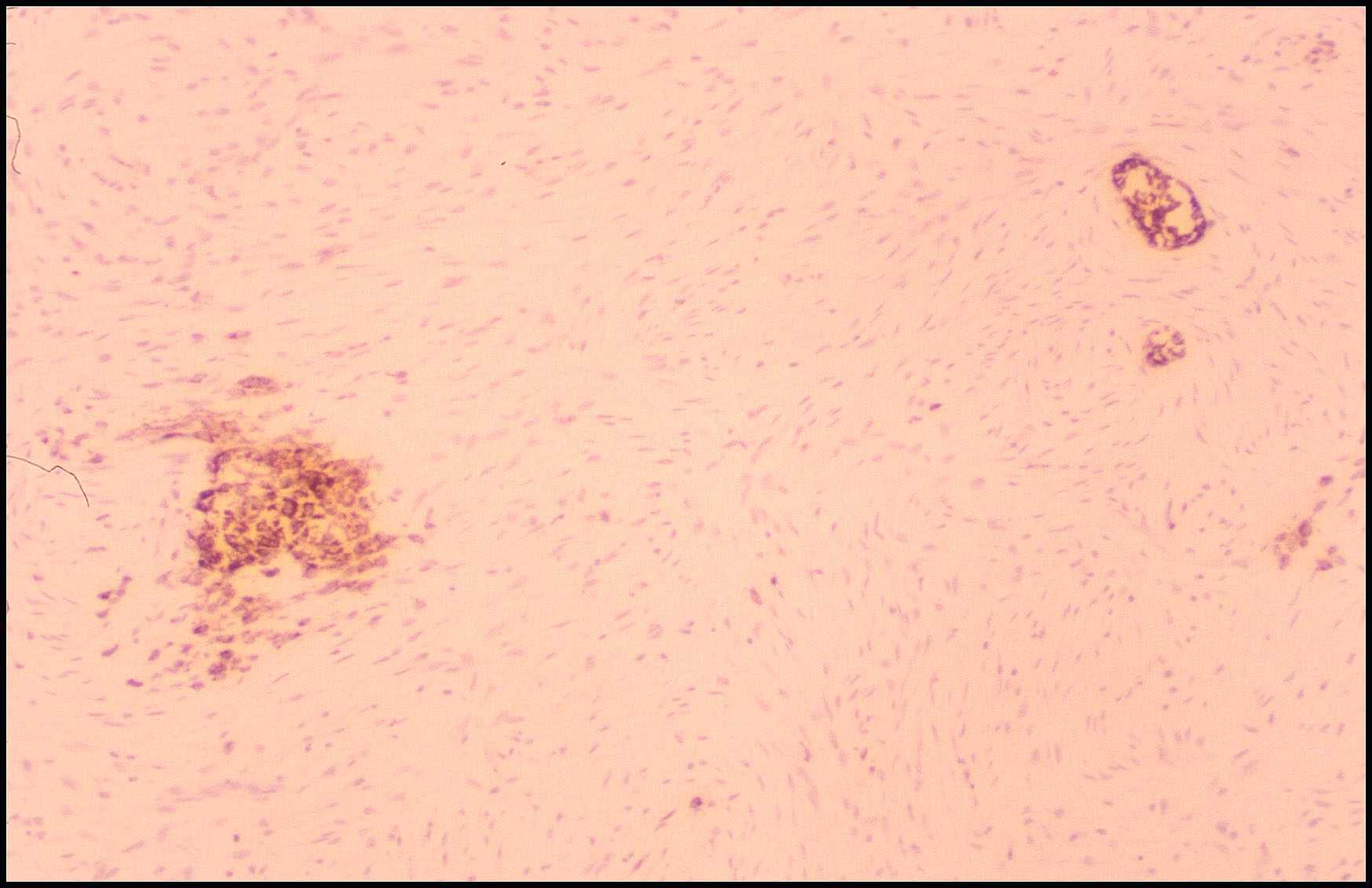
Caso nº 5, del Hospital La Paz. Dr. F. López Barea.- Historia Clínica. Varón de 18 meses de edad. Desde hace varias semanas se le nota una ligera cojera que ha desaparecido cuando acude al Hospital. El estudio radiológico muestra casualmente una lesión intraósea, situada en la metadiáfisis superior del fémur, lítica en la zona periférica y calcificada centralmente; expande al hueso simétricamente. Se procede a un legrado de la lesión e injerto óseo. El paciente se encuentra bien 15 meses después de la intervención. Descripción histológica. La lesión consta de un escaso estroma fibroblástico que diferencia grandes cantidades de estructuras redondeadas, con un centro basófilo y un anillo periférico eosinófilo en un íntimo contacto con las células estromales. El tamaño de estas formaciones es homogéneo, casi acelular, con escasas células incluidas en su seno. La lesión se dispone en sábanas y en la periferia de algunos fragmentos se reconocen pequeñas e irregulares trabéculas óseas, con aislados osteoblastos activos y osteoclastos superficiales y ocasionales osteocitos en lagunas. Con luz polarizada las esférulas calcificadas muestran un patrón reticular de birrefringencia. Diagnóstico anatomopatológico: Lesión fibro-ósea benigna con esférulas calcificadas de un hueso largo (fibroma cementante). Discusión. Un diagnóstico histológico, exclusivamente descriptivo, para esta entidad es el de lesión fibro-ósea benigna con esférulas calcificadas localizada en un hueso largo (1). Esta entidad y en esta situación ha sido referida en la literatura con diferentes nombres: fibroma osificante (2), fibroma cementante (3,4), cementoma (3,5) y displasia fibrosa con "cementículos" (6). Tales denominaciones, que añaden a la morfología una interpretación de la composición del material que diferencia la lesión (hueso de morfología peculiar o cemento), han originado una extrema confusión terminológica a lo que hay que añadir que tales términos son actualmente aplicados a otras lesiones y tumores óseos que se originan en localizaciones específicas. La lesión que aquí se describe es diferente clinicopatológicamente del fibroma osificante de los huesos largos, descrito por Kempson (1966)(7) y actualmente denominado displasia osteofibrosa (8,9). Esta entidad afecta preferentemente al hueso cortical anterior de la diáfisis tibial, en la que suele producir una incurvación anterior. Histológicamente consta de un estroma fibroblástico arremolinado en cuyo seno se diferencian cortas trabéculas óseas revestidas por osteoblastos activos con salpicados osteoclastos. Suele mostrar un fenómeno zonal periférico madurativo. El caso presentado es también diferente clinicopatológicamente al quiste óseo simple juvenil en cuya membrana puede identificarse (15% de los casos) (6,10) focos de un material eosinófilo acelular, que se dispone en masas y gruesas trabéculas, y al que se le denomina " parecido al cemento". La lesión bajo consideración tampoco tiene relación con lesiones sólidas situadas en el cuello y en la diáfisis proximal femoral, de pacientes adultos, que histológicamente contienen grandes cantidades de material "parecido al cemento" y a las que se han denominado "cementomas de los huesos largos" (10).Se interpreta que estas lesiones corresponden a la evolución natural de previos quistes óseos simples juveniles. El aspecto histológico del presente caso sí tiene similaridades morfológicas con la displasia cemento-ósea relacionada con los dientes. Esta es una lesión localizada en los huesos maxilares y relacionada patogénicamente con el ligamento periodontal, estructura que une el diente con el alveolo óseo, y al que se atribuye una capacidad multipotencial de diferenciar tejido conjuntivo, osteoide y cemento (11). No parecería lógico implicar a factores odontogénicos con lesiones óseas alejadas de los maxilares, como es el caso aquí discutido. El fibroma osificante de los huesos de la cara, en especial su variedad psamomatosa, también tiene una importante similitud histológica con la lesión aquí presentada. Este tumor se localiza en la mandíbula, maxilar y en los senos frontales y paranasales (11,12,13,14).Es un tumor que puede tener un comportamiento localmente agresivo y tras se extirpación las recidivas oscilan entre el 20 y el 56% de los casos (12). Brannon y Fowler (2001)(12) sugieren que determinadas localizaciones (fosa nasal y proximidad de la base anterior del cráneo) que dificultan su abordaje quirúrgico son la causa de sus frecuentes recidivas lo que puede obligar a un tratamiento quirúrgico más radical que un simple legrado incompleto. La lesión aquí discutida, situada en un hueso largo, ha sido referida en la literatura muy infrecuentemente. Black y cols (1991)(4) refieren sólo 4 casos previos (2,3,5) y describen uno nuevo. Sissons y cols (1993)(1) estudian, desde un punto de vista exclusivamente histológico, 20 lesiones fibro-óseas con esférulas calcificadas distribuidas por todo el esqueleto e identifican 3 nuevos pacientes (Casos 3,13 14) con un lesión en un hueso largo que carece de evidencia clinicopatológica de displasia fibrosa. Incluyen en este estudio 2 casos previamente publicados de esta entidad (Casos 1 y 2)(2). La Tabla 1 resume las características clinicopatológicas de los 8 pacientes revisados en la literatura y el caso aquí discutido, de lesiones fibro-óseas benignas con esférulas calcificadas localizadas en los huesos largos. Tabla 1. Características clinicopatológicas de 9 pacientes con lesión fibro-ósea con esférulas calcificadas de los huesos largos
Se trata de 5 hombres y 4 mujeres, con una edad media de 21 años y con edades extremas de 18 meses y 53 años. Las lesiones se localizan en la tibia (5) y 1 en fémur, húmero, peroné y metacarpiano respectivamente. Siete lesiones son diafisarias, 1 metaepifisaria y 1 metafisaria. Radiológicamente todas las lesiones son líticas, 5 con calcificaciones centrales y 7 son expansivas. Histológicamente las lesiones se caracterizan por un estroma fibroblástico denso compuesto por células fusiformes, con núcleos vesiculosos, regulares y sin nucleolo. La actividad mitósica es baja. En su seno se diferencian numerosas estructuras globulares, de tamaños variables entre 20 y 60 micras. El centro de las esférulas suele ser basófilo y su periferia eosinófila. Este material es casi acelular pero ocasionalmente se identifican células en su interior. Con luz polarizada se evidencia un patrón reticular de las fibras de colágena. En 3 casos (Casos 2 y 3 de Sissons y cols (1) y el caso aquí presentado) la lesión se acompaña de ocasionales trabéculas óseas, cortas e irregulares en su forma, con aislados osteoblastos cúbicos y osteoclastos en su periferia y osteocitos en su interior. Dos casos (Caso 3 de Sisson y cols (1) y el caso de Horn y cols (5)) han sido estudiados con microscopía electrónica. El componente celular de la lesión muestra caracteres ultraestructurales de fibroblastos y posibles miofibroblastos situados en el seno de abundantes fibras de colágena. Estas se condensan y ordenan circularmente alrededor de las esférulas cuyo centro se encuentra altamente calcificado. Existe constancia del tratamiento en 6 pacientes en los que se realizó un legrado de la lesión e injerto de hueso. Se conoce el seguimiento de 4 de ellos que no mostraron recidiva en un periódo de 15 meses, 2 , 4 y 5 años. Ya ha quedado expresada previamente la diferente interpretación nosológica, y por ello la variada nomenclatura, aplicada a la lesión fibro-ósea con esférulas calcificadas. Basado en la localización de esta lesión, no sólo descrita en los huesos largos sino también en vértebras, iliaco, y en los huesos de la cara excluidos los maxilares (1,11,13,14), regiones sin relación con el ligamento periodontal, unido a los hallazgos ultraestructurales que asemejan a esta lesión a la displasia fibrosa (1,15), podría concluirse que las esférulas calcificadas son una forma peculiar de hueso y no auténtico cemento. Además, la existencia de típicas displasias fibrosas con ocasionales esférulas calcificadas ( 1,16), lesiones fibro-óseas con numerosas esférulas calcificadas que se acompañan de pequeñas e irregulares trabéculas óseas diferentes de las de la displasia fibrosa, y lesiones fibro-óseas compuestas exclusivamente de esférulas calcificadas, hacen sugerir a algunos autores (1,16) que todas ellas pertenecen a un amplio espectro en uno de cuyos extremos se sitúa la típica displasia fibrosa y en el otro la lesión fibro-ósea con esférulas calcificadas (fibroma cemento-osificante para Voyteck y cols (1995))(16). No obstante, no se debe olvidar, que similares lesiones histológicas a la de la lesión fibro-ósea con esférulas calcificadas, y localizadas en los huesos de la cara (fibroma osificante, variedad psamomatosa)(1,11,12,13,14), pueden tener un comportamiento localmente agresivo. Sería deseable dar a conocer un mayor número de casos de lesiones fibro-óseas con esférulas calcificadas localizadas en los huesos largos para confirmar que su comportamiento evolutivo es menos agresivo, como parece intuirse en esta corta casuística. Bibliografía. 1. Sissons HA, Steiner GC, Dorfman HD. Calcified spherules in fibro-osseous lesions of bone. Arch Pathol Lab Med 1993; 117: 284-290. 2. Sissons HA, Kancherla PL, Lehman WB. Ossifying fibroma of bone. Report of two cases. Bull Hosp Joint Dis 1983; 43: 1-14. 3. Kolár JJ, Horn V, Zidková H, Sprindrich J. Cementifying fibroma (so-called "cementoma") of tibia. Br J Radiol 1981; 54: 989-992. 4. Black DL, De Smet AA, Neff JR, Bhatia P. Case report 695: Cementifying fibroma of the proximal end of the tibia. Skeletal Radiol 1991; 20: 543-546. 5. Horn V, Bozdech Z, Macek M, Foukal T, Zidková H. Cementoma-like tumours of bone. Arch Orthop Trauma Surg 1982; 100: 267-272. 6. Mirra JM, Gold RH. Fibrous dysplasia. En Bone Tumors. Clinical, radiologic,and pathologic correlations. Ed. Mirra JM. Lea & Febiger, Philadelphia 1989: 191-226. 7. Kempson RL. Ossifying fibroma of the long bones: a light and electron microscopic study. Arch Pathol 1966; 82: 218-233. 8. Campanacci M, Laus M. Osteofibrous dysplasia of the tibia and fibula. J Bone Joint Surg (A) 1981; 63: 367-375. 9. Park Y-K, Unni KK, McLeod RA, Pritchard DJ. Osteofibrous dysplasia: clinicopathologic study of 80 cases. Hum Pathol 1993; 24: 1339-1347. 10.Adler CP. Tumour-like lesions in the femur with cementum-like material. Does a " cementoma" of long bone exist?. Skeletal Radiol 1985; 14: 26-37. 11.Margo CE, Radgsdale BD, Perman KI, Zimmerman LE, Sweet DE. Psammomatoid (juvenil) ossifying fibroma of the orbit. Ophthalmology 1985; 92: 150-159. 12.Brannon RB, Fowler CB. Benign fibro-osseous lesions: a review of current concepts. Adv Anat Pathol 2001; 8: 126-143. 13.Slootweg PJ, Panders AK, Koopmans R, Nikkels PGJ. Juvenile ossifying fibroma. An analysis of 33 cases with emphasis on histopathological aspects. J Oral Pathol Med 1994; 23: 385-388. 14.El-Mofty S. Psammomatoid and trabecular juvenile ossifying fibroma of the craniofacial skeleton: two distinct clinicopathologic entities. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2002; 93: 296-304. 15.Greco MA, Steiner GC. Ultrastructure of fibrous dysplasia of bone: a study of its fibrous, osseous and cartilaginous components. Ultrastruct Pathol 1986; 10: 55-66. 16.Voytek TM, Ro JY, Edeiken J, Ayala AG. Fibrous dysplasia and cemento-ossifying fibroma. A histologic spectrum. Am J Surg Pathol 1995; 19: 775-781.
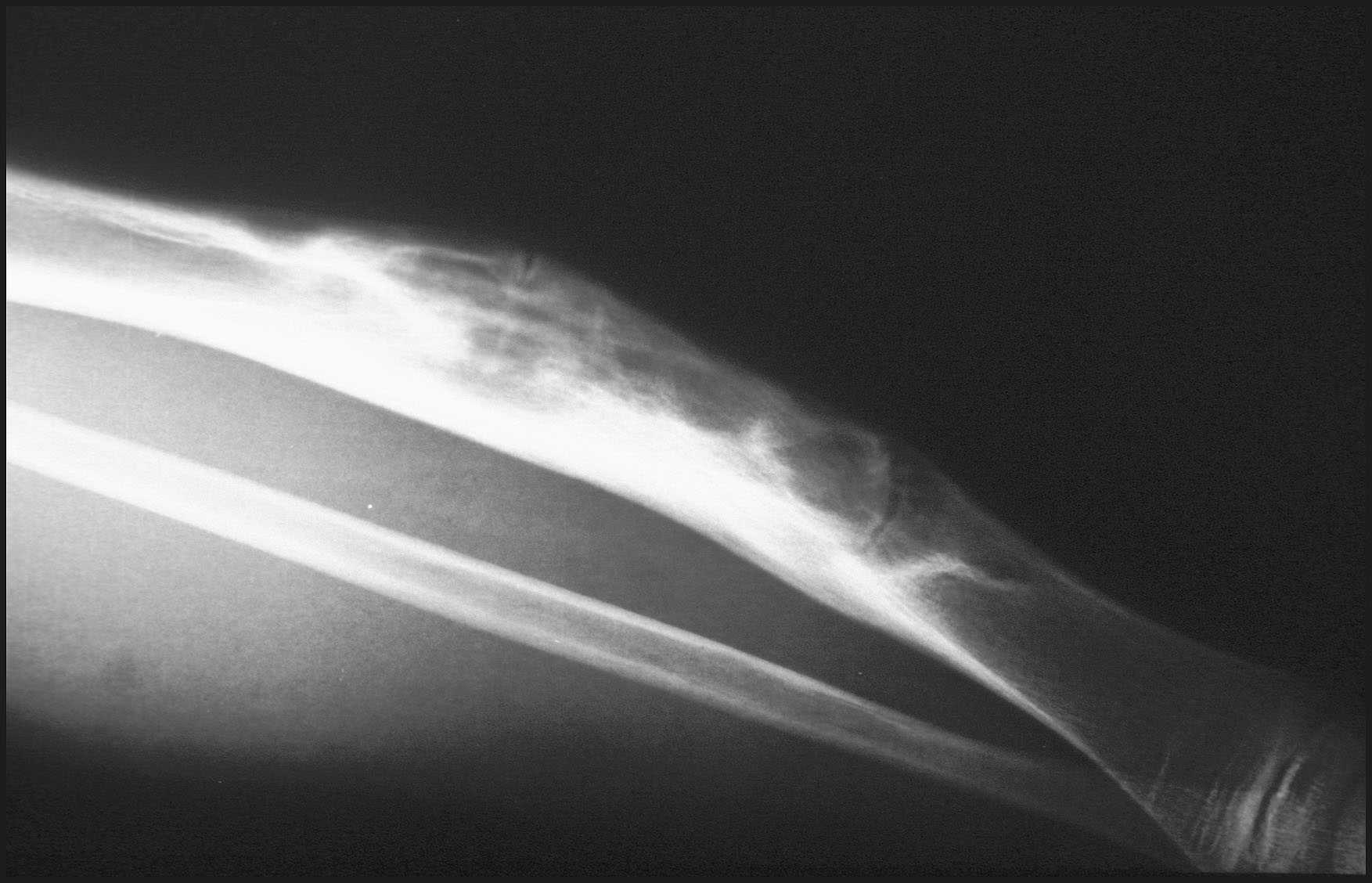
CAS0 Nº6.- Dra. I. González Mediero. Hospital. Niño Jesús. Madrid. Resumen de Caso: Niño varón de 6 años, que a los 3 años muestra molestias y lesión en tibia dcha, que se biopsia y se diagnostica de Displasia Fibrosa en otro centro. Los síntomas han ido aumentando con incurvación anterior de la tibia, observándose radiológicamente una extensa lesión diafisaria anterior lítica y multifocal con bordes esclerosos y predominantemente intracortical. La lesión es resecada y cureteada en su base, rellenándose y fijándose con Ortofix. Histológicamente se observa una lesión fibrosa con estroma más ó menos denso, con un patrón interpuesto de trabéculas con reconocible ribete osteoblástico activo periférico. En el estudio inmunohistoquímico se observa positividad estromal para la Vimentina, y ocasionales agrupamientos de escasas células que expresan citoqueratina, AE1-3. DIAGNÓSTICO: "DISPLASIA OSTEOFIBROSA ADAMANTINOMATOSA".Versus "ADAMANTINOMA JUVENIL tipo D. OF. (D. OF (A) / A.J (D. OF). DISCUSIÓN: La D.OF (A), o AJ (D.OF), es una lesión benigna poco frecuente, localizada en tibia y peroné en la infancia y caracterizada por tejido fibroso y trabéculas de hueso inmaduro no lamelar, con ribete periférico de osteoblastos activos (1). Forma parte de las lesiones pseudotumorales del hueso, junto con otras, como la Displasia Fibrosa, Fibroma no Osificante (Defecto Fibroso Metafisario), Displasia Fibrocartilaginosa, y otras (2). Como todas ellas ha sufrido cambios en la nomenclatura. En 1921 Frangehenhein hizo la primera publicación con el nombre de "Osteoitis Fibrosa Congénita", seguido entre otras por el de "Fibroma Osificante de los huesos largos" (3), "Displasia Fibrosa Intracortical" (3), "Displasia Fibrosa de Tibia y Peroné" (4), variante de Displasia Fibrosa (5), y a partir de los años 90 Sweet y Campanacci en 1992 y 94 (3, 6), y Kuruvilla y Steiner en 1998 (7), (10) refieren la relación de la Displasia Osteofibrosa y el Adamantinoma, mediantes sus estudios con inmunohistoquímica. El origen de ésta lesión también es causa de distintas controversias. Por su edad de aparición en los primeros diez años, su progresión hasta la detención del crecimiento del esqueleto, y su frecuente recidiva en dicha edad después de la resección, se ha considerado que dicho comportamiento era propio de las proliferaciones mesenquimales congénitas y hamartomatosas (4). Con respecto a la relación entre el Adamantinoma y la D.OF, se ha referido que son distintas expresiones del mismo proceso (3); que es una lesión regresiva del Adamantinoma ó Adamantinoma Diferenciado de la clasificiación de Czerniak y Dorfman (7, 9), así como precursor o residuo del mismo (8). Han sido sobre todo los estudios con la inmunohistoquímica, los que han planteado éstas últimas opiniones y la expresión de la Citoqueratina, AE1-3 en grupos, (menos de 12 células) y aisladamente en el estroma, así como su patrón fibroso y trabecular reticular, lo que ha inducido a la denominación de D. OF Adamantinomatosa, Adamantinoma juvenil tipo D.OF ó Adamantinoma Diferenciado (3, 7, 9) y a sugerir un origen común en ambas (6). El Diagnóstico Diferencial debe hacerse sobre todo con el Adamantinoma Clásico, cuya aparición se halla por encima de los 20 años, su localización es diafisaria en tibia y peroné, la lesión es lítica, excéntrica aunque puede extenderse al periostio y la expresión de la AE1-AE3 se expresa en grandes agrupamientos y distintos patrones (7, 8, 9). Otros diagnósticos a tener en cuenta por sus anteriores referencias, es con la Displasia Fibrosa, cuya clínica, radiología e histología son distintas., con lesión uni o multifocal, en cualquier localización, intramedular y con predominio fibroso e interposición de trabéculas exentas de ribete osteoblástico, en el patrón clásico. (1, 2). El tratamiento de la D.OF Adamantinomatosa, debe ser resección en bloque, con cureteado, por su posible recidiva y su potencial comportamiento agresivo. (9). Bibliografía: Schajovicz F. Histological typing of Bone Tumours WHO. 1993. P. 40. Helliwel T. Pathology of Bone and Joint Neoplasms. MP. Saunders Company. 1999. 266. Sweet DE, Vinh TN and Devanes K. Cortical Osteofibrous Dyplasia of Long Bone and its Relationshin to Adamantinoma. A Clinicopathologic Study of 30 cases. Am J Surg Pathol. 1992. 16. 3. 282-90. Camapacci M, and Laus M. Osteofibrous Dysplasia of the tibia and Fibula. J. Bone and Joing Surg. 1981. 63. 3. 367-375. Campbell CJ, Hawk T. A variant of fibrous dysplasia (Ostefibrous displasia) J. Bone Joint Surg 64 A: 231-236. 1982. Benassi MS, Campanacci L, Gamberi G, Ferrari C, Picci P, Sangiorgi L and Campanacci M. Cytokeratin exprexion and distribution in adamantinoma of the long bonrs and osteofibrous dysplasia of tibia and fibula. An inmunohistochemical study con correlated to histogeneris. Histopatology 1994. 25. 71-76. Kuruvilla G, and Steiner G. Osteofibrous Dysplasia - like Adamantinoma of Bone: A Report of five cases with Inmunohistochemical and. Ultrastructural Studies. Hum Pathol. 1988. 28. 8. 809-814. Springfield D, Rosenberg A, Mankin HJ, and Mindell ER. Relationship Between Osteofibrous Dyplasia and Adamantinoma. Clin Orthop R.R. 1994. 309. 234-244. Czerniak B, Rojas- Corona R. and Dorfman. Morphologic Diversity of Long Bone Adamantinoma. The Concept of Differentiated (Regressin). Adamantinoma and irs Relation ship to Osteofibrous Displasia. Cancer 1989. 64. 2319-2334. Fletcher D. M., Unni K., Mertres F. Pathology and Genetics. Tumours of Soft Tissue and Bone. WHO. Classification of tumorers. IARC Press Lyon 2002.
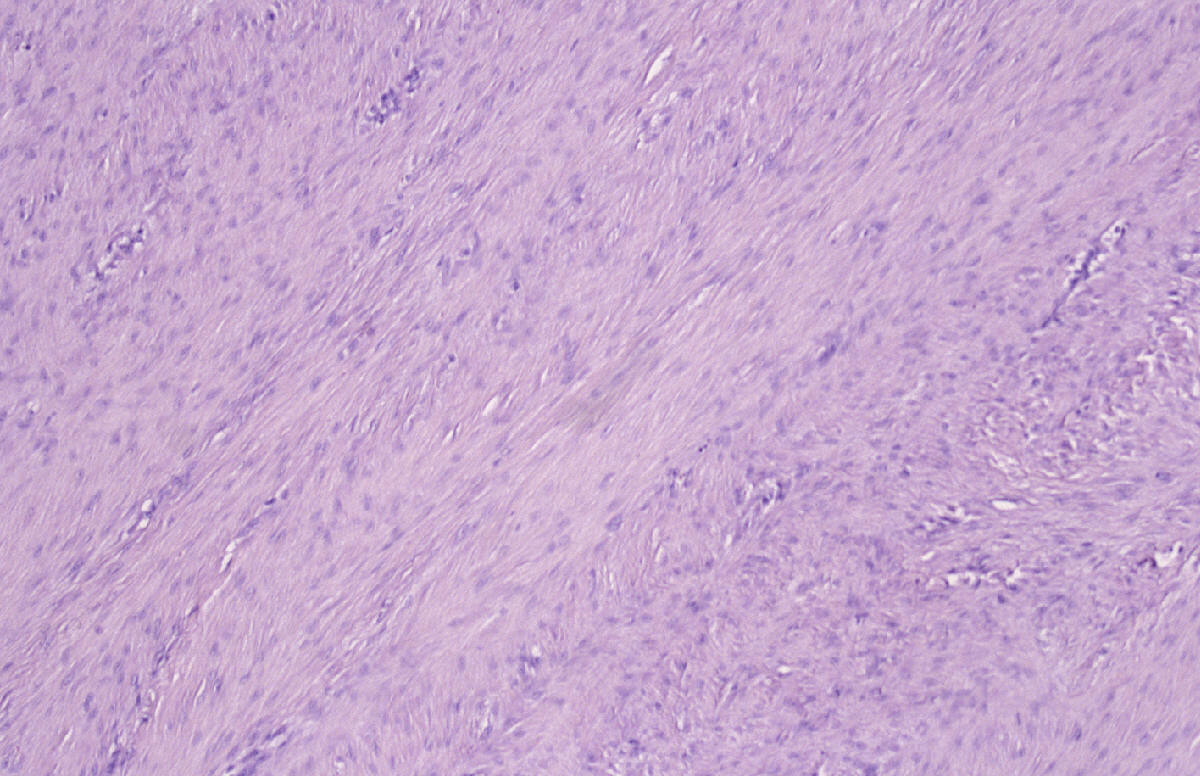
CASO Nº7 Dr. Vila Torres Paciente varón que a los 6 meses de edad presentaba una deformidad en pierna izquierda. Tras estudio radiológico y biopsia en otro centro, fue diagnosticado de "Lipomatosis focal parafibular". A la edad de 1 año, ingresó en otro centro para revisión de la deformidad en pierna izquierda cuyo estudio radiológico demostró una incurvación del peroné producida por una tumoración de partes blandas que se extirpó. A los 2.5 años de edad acude a nuestro centro por recidiva de la tumoración, practicándose resección en bloque del peroné. La laminilla pertenece a la tumoración resecada. DISCUSIÓN Las secciones histológicas muestran una proliferación de células fusiformes dispuestas en haces, mostrando mitosis ocasionales y, focalmente, zonas de fibrosis colágena. No se observa necrosis. En las secciones óseas, se observa un patrón permeativo en el termio medio, en relación con la zona de seudoartrosis. Periféricamente, se identifica tejido muscular esquelético, infiltrado por la proliferación fibroblástica. La IHQ mostró expresión intensa de vimentina, mientras que no se demostró expresión de actina, desmina, Myo-D1, CD34, S-100 ni de AE1/AE3. DIAGNÓSTICO. Fibromatosis infantil del tipo desmoide, con afectación del peroné y partes blandas. La fibromatosis infantil de tipo desmoide, es una variante de fibromatosis agresiva. Stout definió el concepto de fibromatosis agresiva, como una proliferación fibroblástica infiltrante de potencial biológico incierto, focalmente agresiva y no metastatizante. Invade músculo, tejido subcutáneo y estructuras neurovasculares, pero la invasión de hueso es rara. Morfológicamente está constituída por una proliferación de fibroblastos de aspecto benigno. Como otros tumores de tipo desmoide, el curso natural es impredecible: agresivo, estacionario o remisión espontánea. El tratamiento debe ser ser, siempre que sea posible, una resección amplia con márgenes libres (la RNM preoperatoria y la biopsia peroperatoria ayudan a identificar unos márgenes libres). En casos irresecables, se puede intentar quimioterapia y tratamiento hormonal aunque no existen unos criterios bien definidos sobre su utilización. También se ha indicado la radioterapia como último recurso por la posibilidad de desarrolar un segundo tumor. El diagnóstico diferencial incluye otros tumores fibrosos infantiles: Fibromatosis desmoide, Miofibromatosis juvenil, Fibrosarcoma congénito-infantil, Fibrosarcoma tipo adulto, Fibromatosis colli, Hamartoma fibroso infantil, Angiofibroma, Fibromatosis hialina, Fibromatosis digital infantil, Tumores y lesiones reactivas de la superficie del hueso, Miositis osificante y Fibroma desmoplástico. En nuestro caso llama la atención la asociación de una fibromatosis de tipo desmoide con afectación simultánea de partes blandas y de hueso. BIBLIOGRAFIA H.D Dorfman, B. Czerniak Bone Tumors Mosby Inc. 1998, S. Louis Missouri, USA Dahlin´s Bone Tumors K.K. Unni Lippincot-Raven , Philadephia, 1996 Small round cell tumors of bone and soft tissue Llombart-Bosch. En Seminars in Diagnostic Pathology, 13/3(153-170)-1996 D.J. Santacruz. Ed.
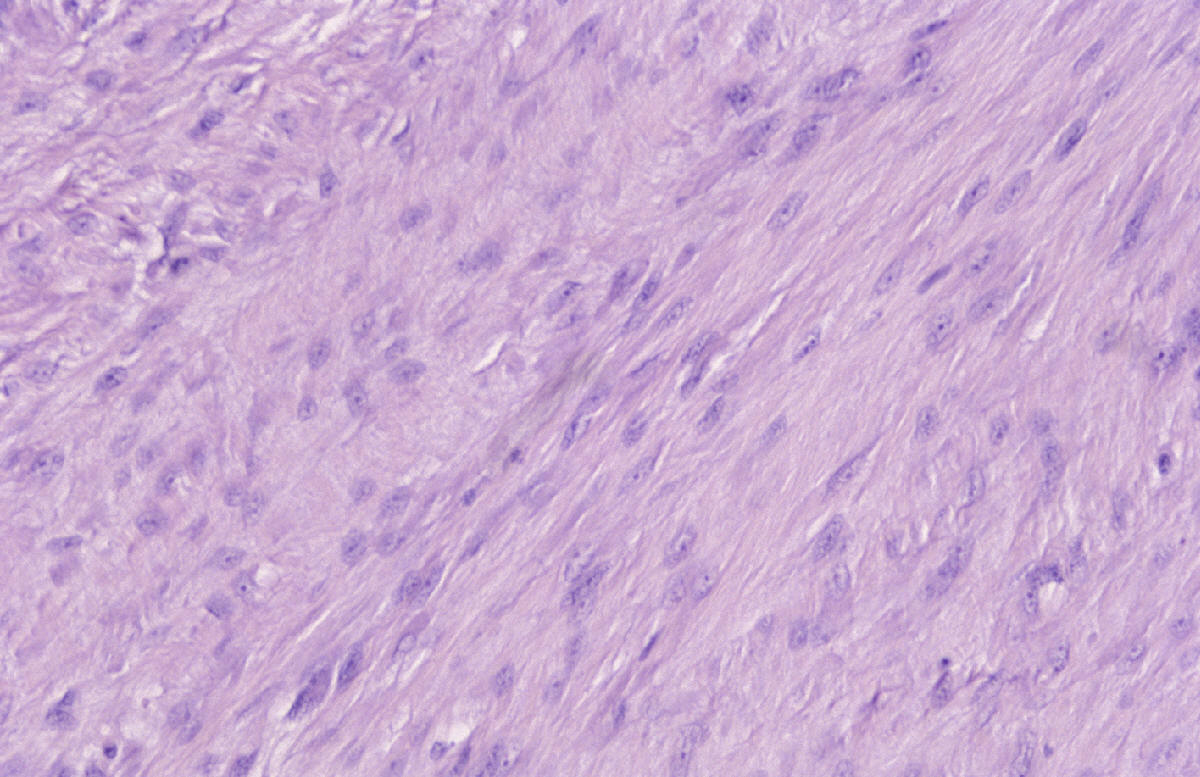
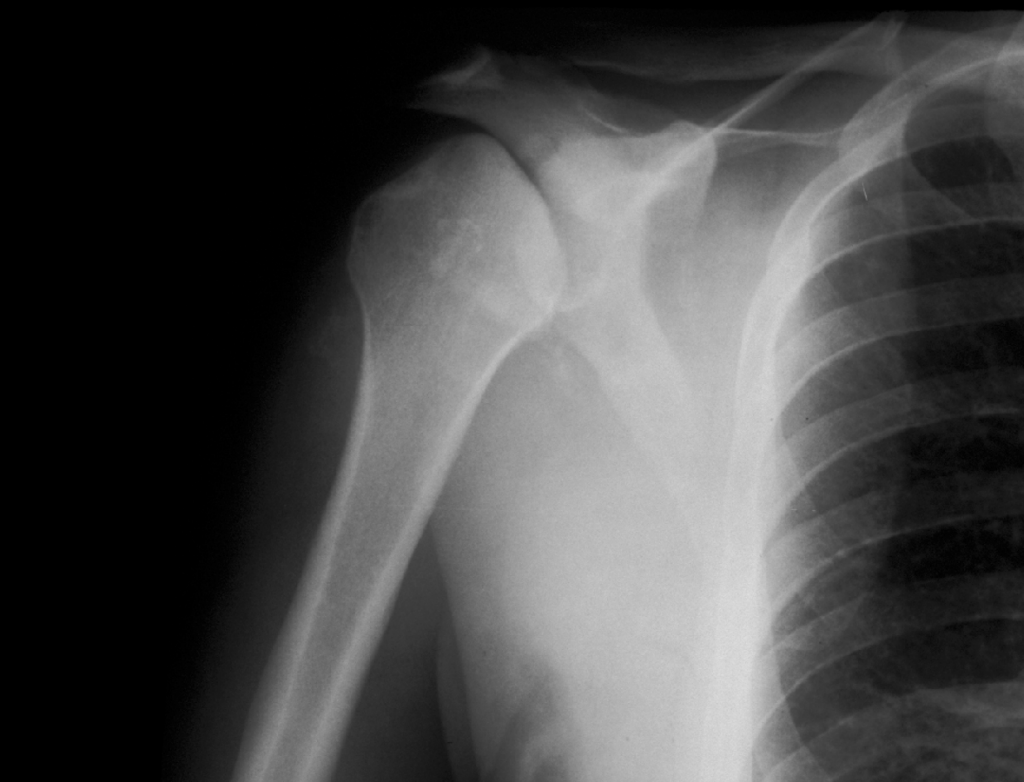
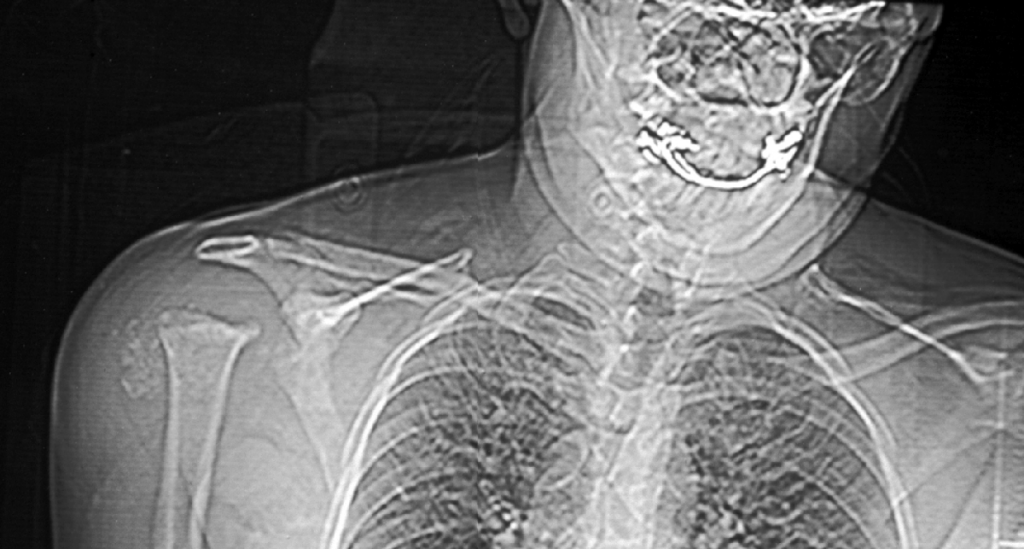
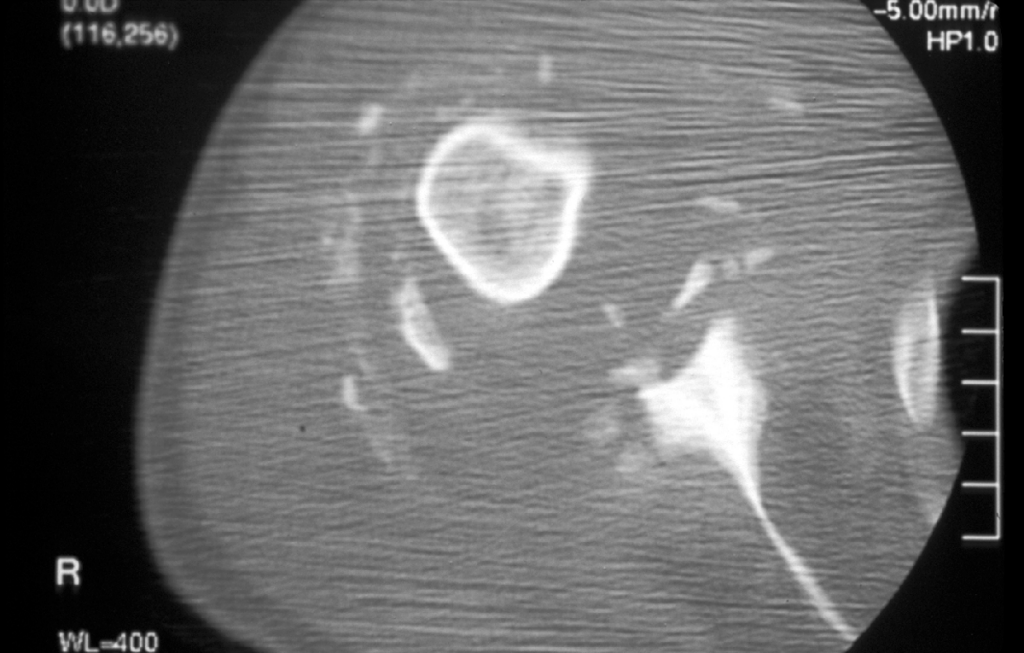
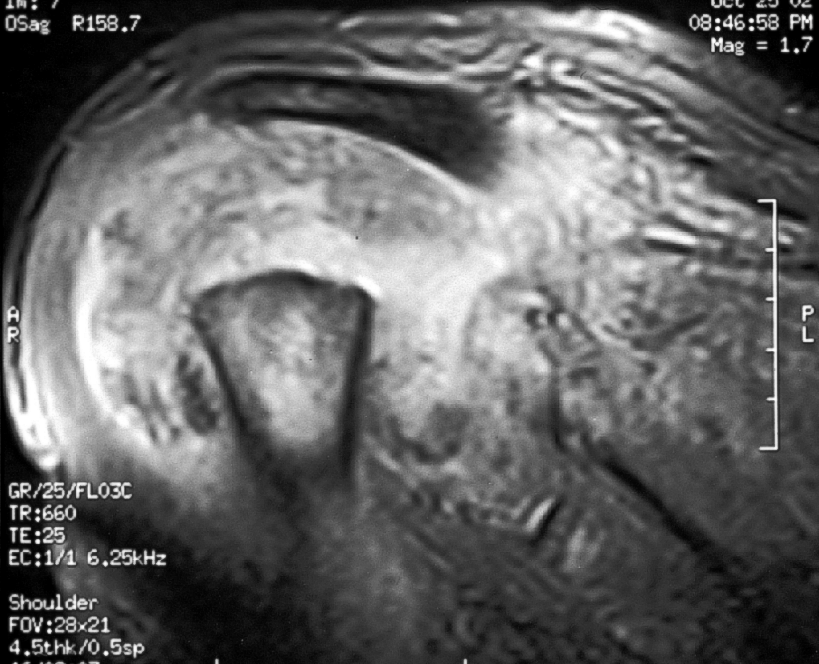
Caso nº 8. FJ Martínez Tello. Osteoartritis rápidamente destructiva. Historia clínica. Varón de 47 años de edad con historia de hombro doloroso derecho de un año de evolución que no cede al tratamiento médico ni a rehabilitación habituales. Radiológicamnete sin hallazgos de interés hasta hace aproximadamente un mes, cuando se pone de manifiesto una osteolisis masiva de la cabeza del húmero, desestructuración de la cavidad articular glenohumeral y un marcado aumento del espacio subacromial junto a alteración marcada de las estructuras musculares. Diagnóstico clínico : Sarcoma sinovial. Discusión. El caso presentado es un raro ejemplo de osteoartritis ( artrosis) rápidamente destructiva ( RDO: Rapid Destructive Osteoartritis ) de la cabeza humeral, cavidad glenoidea y acromion ya que la localización más típica de este proceso, y donde primero fue descrita por Forestier en 1957, es en la cadera, habiéndose denominado Rapid Destructive Hip Disease ( RDHD ), esto es, enfermedad de la cadera rápidamente destructiva. Se trata de un proceso muy infrecuente que se caracteriza clínicamente por : 1- Rápida destrucción de la artiulación ( promedio menos de un año ) ; 2- mayor afectación del sexo femenino; 3- producirse en edads más avanzadas ( promedio 72 años, superior a la de lartrosis común ) ; 4 preservación relativa del rango de movimientos. Anatomopatológicamente sólo se hallan graves cambios degenerativos, con leve respuesta inflamatoria crónica sinovial, no tan intensa como para sugerir una artritis inflamatoria, ausencia de osteonecrosis u osteonecrosis segmentaria en la región subarticular, consistente con osteonecrosis secundaria y no primaria, y ausencia de osteofitos. La causa no es conocida pero se ha postulado que se trate de una rara variante de artrosis de evolución rápidamente progresiva. Tiene similitudes clínicas con varias entidades cuya etiología se ha tratado de imputar pero que no ha sido demostrada y que hay que tener en cuenta en el diagnóstico diferencial: 1-La Artropatía inducida por cristales, cómo la de la condrocalcinosis, la asociada a apatita, y el cuadro del "hombro de Milwaukee" ( Milwaukee shoulder) en el que microesferas que contienen cristales de hidroxiapatita producen desorganización de la articulación del hombro tras la rotura de la vaina del rotador y liberación de cristales en la articulación. 2- Necrosis isquémica; 3-Artropatía iatrogénica inducida por fármacos ( analgésicos ), especialmente la Indometacina; y de mayor importancia 4- la neuroartropatía ( tabes, siringomielia) y 5- la artritis séptica, que deben excluirse clínicamente, ya que en ambas se obtienen pésimos resultados con la implantación de una prótesis, en tanto que en la RDO los resultados han sido buenos. Para realizar el diagnóstico de RDO es fundamental el estudio del líquido sinovial para excluir otras causas. Literatura: 1- Forestier F. Coxite rhumatismales subaigues et chroniques. Tésis. Paris. 1957. 2- Rosenberg ZS, Shankman S, Steiner GC, Kastenbaum DK, Norman A, Lazansky MG. Rapid Destructive Osteoarthritis: Clinical, Radiographic and Pathological Features. Radiology 182: 213-216, 1992. 3- Bock GW, García A, Weisman MH, Mjor PA, Lyttle D, Haghighi P, Greenway GD, Resnick D. Radiology 186: 461-466, 1993. 4- Lawrance JAL, Athanasou NA. Rapidly destructive hip disease. Skeletal Radiol 24: 639-641, 1995.
[Programa] [Cursos Cortos] [Seminarios]
|
. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| © SEAP. Sociedad Española de Anatomía Patológica | Actualizado: 09/07/2003 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||