|
Información || Congresos || Cursos || Territoriales || Noticias || Patología || Telepatología |
|
Información || Congresos || Cursos || Territoriales || Noticias || Patología || Telepatología |
| . |
[XXII Congreso Nacional (Palma de Mallorca)] [XXI Congreso Nacional (Madrid)] [ XX Congreso Nacional (Pamplona) ] [XIX Congreso Nacional (Barcelona) ] [XVIII Congreso Nacional (Málaga) ]
SEMINARIO DE NEFROPATOLOGÍA MARIA JOSE PANADES SIURANA RESUMEN CLINICO Mujer de 34 años, sin antecedentes familiares ni personales de interés. Historia actual: -febrícula y anemia de trastornos crónicos de 1 año de evolución. -3 meses antes del ingreso: edemas progresivos y anasarca. -analítica: proteinuria de 9 grs/24 hores, Bence-Jones (-) función renal normal. -DIAGNOSTICO CLINICO: SINDROME NEFROTICO FLORIDO Se practica biopsia renal. (B/02/5637) Durante la realización de la biopsia renal por control ecográfico, se detecta casualmente una masa retroperitoneal. Se realiza posteriormente TAC abdominal, observándose una masa localizada a nivel epigástrico, tronco-celíaco, que desplaza el cuerpo del pancreas. El TAC torácico fue normal. Se extirpa la masa posteriormente, (B/02/5816), al cabo de 15 dias de realizar la biopsia renal.
HALLAZGOS MACROSCOPICOS BIOPSIA RENAL: Varios cilindros de tejido, el mayor de 1 cm de longitud. MASA RETROPERITONEAL: Fragmento de 6x4 cms de tamaño, de coloración blanquecina y consistencia blanda, sin observarse zonas de aspecto necrótico-hemorrágico.
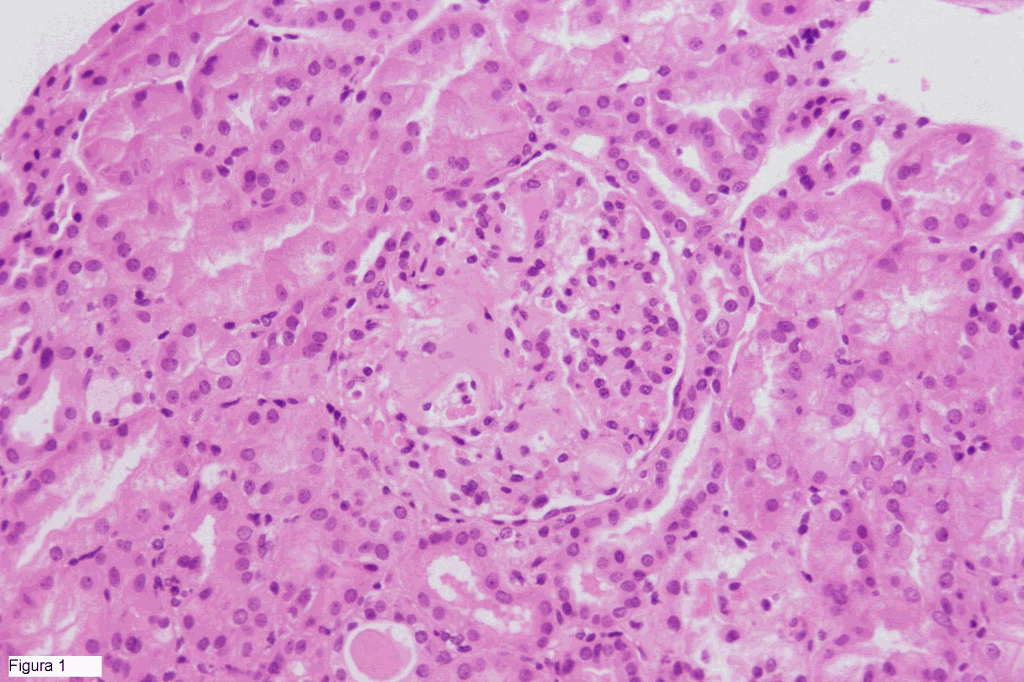
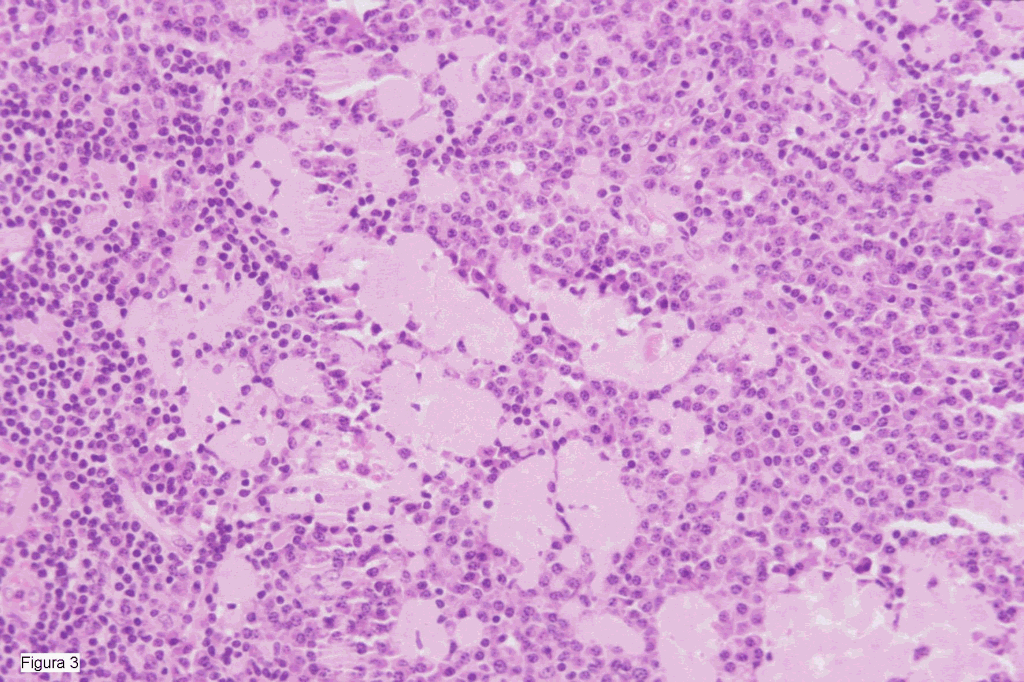
HALLAZGOS MICROSCOPICOS Los fragmentos de tejido renal (figura 1) mostraban unos glomérulos con depósitos de material amiloide, Rojo Congo positivo y con birrefringencia verdosa. Las paredes vasculares tambien presentaban depósitos amiloides. Inmunofluorescencia negativa. La masa retroperitoneal (figuras 2 i 3) correspondia a parenquima linfoide mostrando una hiperplasia de los folículos, algunos con estructuras hialino-vasculares y rodeados por linfocitos adoptando una morfologia en "capas de cebolla". El espacio interfolicular mostraba un marcado infiltrado por células plasmáticas maduras, junto a abundante estroma fibrohialino. La tinción de Rojo Congo demostró importantes depósitos de material amiloide a nivel de los vasos y del estroma fibrohialino. La tipificación inmunohistoquímica con anticuerpos contra la proteina AA, fue intensamente positiva.
DIAGNOSTICOS: ENFERMEDAD DE CASTLEMAN, DE TIPO MIXTO, CON AMILOIDOSIS RENAL.
COMENTARIO Las complicaciones renales de la enfermedad de Castleman(E.C) son poco frecuentes, entre las cuales se incluyen la enfermedad por cambios mínimos, glomerulonefritis mesangiocapilar, membranosa y proliferativa mesangial. (1) La amiloidosis sistémica es una complicación muy rara de la E.C. En la literatura, hasta el año 2002 , solo se han publicado 20 casos, de los cuales solo 11 presentaban síndrome nefrótico causado por amiloidosis renal. (2,3). La forma localizada de la E.C. afecta principalmente al mediastino, siendo la localización peritoneal o retroperitoneal mucho mas rara. La forma multicéntrica presenta una evolucion mas agresiva y suele ser la variante de células plasmáticas. El tipo hialino-vascular es mucho mas frecuente y en ocasiones, formas mixtas se han descrito, como el caso que presentamos. El principal tipo de amiloide depositado en los casos descritos y en el nuestro es el AA. Aunque no se conoce el mecanismo exacto de producción, en los últimos trabajos parece jugar un papel importante la interleukina-6 (IL-6) en la patogénesis del depósito de amiloide en la E.C. (4) El tratamiento de la E.C. y la posible desaparición de la amiloidosis y síndrome nefrótico es muy controvertido y basado principalmente en los casos publicados. En algunos casos, desaparece la amiloidosis y el síndrome nefrótico despues de extirpar el ganglio linfático (5). En otros, disminuye el síndrome nefrótico después de extirpar el tumor, pero los depósitos de amiloide en riñón persisten un año después de la cirugia. (4). Sin embargo, en algunos casos, no disminuye el síndrome nefrótico (6) y en otros, disminuye después de administrar Colchicina (7). Nuestro caso presentó una evolución desfavorable. Después de la extirpación del tumor, la paciente continuó con un síndrome nefrótico florido (12 grs/24h) y con una función renal normal hasta la actualidad (creatinina: 08 mg/dl). No ha recibido tratamiento y actualmente está gestante de 10 semanas.
BIBLIOGRAFIA:
[Programa del Congreso] [Índice del Seminario] [Cursos Cortos] [Seminarios]
|
. | |||
| © SEAP. Sociedad Española de Anatomía Patológica | Actualizado: 09/07/2003 |
||||