
T.7
- AVANCES EN EL DIAGNÓSTICO Y TRATAMIENTO DE LA AMILOIDOSIS PRIMARIA
Dr.
Joan Bladé. Institut de Ma1alties Hematooncológiques. Servei
d'Hematologia. Institut d’Investigacions Biomèdiques August Pi i Sunyer
(IDIBAPS). Hospital
Clínic. Barcelona.
GENERALIDADES
La amiloidosis es un trastorno caracterizado
por el depósito de una substancia amorfa en diversos órganos y tejidos. Bajo la
luz polarizada, la substancia amiloide se tiñe con el rojo Congo y produce una
birrefringencia de color verde manzana. La substancia amiloide está compuesta
por una red de fibrillas unidas entre sí. Estas fibrillas están dispuestas en
antiparalelo (estructura beta plegada). El plegamiento en disposición beta de
las fibrillas es la responsable de las características tintoriales y de las
propiedades ópticas de la sustancia amiloide. Todas las formas de amiloidosis
son rojo Congo positivas y lo que determina el tipo de amiloidosis es el
estudio inmunohistoquímico de la sustancia amiloide.
La sustancia amiloide es rica en
gIicosaminoglican, substancia que puede tener propiedades fibrinogenésicas
(1,2). Otro componente constante de los depósitos amiloides es el denominado
componente P, una glicoproteína resistente a las proteinasas que también se
encuentra en el suero (SAP). La posibilidad de emplear técnicas gammagráficas
utilizando SAP marcado con radioisótopos ha proporcionado información valiosa
sobre la historia natural y la respuesta al tratamiento en la amiloidosis
sistémica. De hecho, la disminución de la producción de la proteína precursora
de la fibrilla amiloide se ha correlacionado con la disminución del depósito de
substancia amiloide. Ello sugiere que la amiloidosis es un proceso más dinámico
de lo que se había considerado (2).
En todas las formas de amiloidosis, la
formación y depósito de las fibrillas amiloides precisan de la existencia de la
respectiva proteína precursora. Las razones por las cuales el material amiloide
se deposita en unos individuos y no en otros, los factores que determinan la
distribución anatómica del depósito, la velocidad de comienzo y progresión, así
como las distintas consecuencias clínicas del depósito amiloide no están
plenamente establecidos (1,2).
En este curso revisaremos las características
clínicas, el pronóstico y el tratamiento de la amiloidosis primaria.
AMILOIDOSIS
PRIMARIA
Concepto e incidencia
En la amiloidosis primaria (AL) las fibrillas
amiloides están compuestas por la porción variable de una cadena ligera. La
cadena ligera es con mayor frecuencia lambda que kappa (relación 3:1). La edad
mediana al diagnóstico se sitúa alrededor de los 65 años. La incidencia anual
se cifra en alrededor de 8 casos nuevos por millón de habitantes y año (3).
Manifestaciones clínicas y de laboratorio
El cansancio y la pérdida de
peso, constituyen los síntomas más frecuentes de la AL (4,5). Los pacientes que
tienen insuficiencia cardíaca suelen presentar disnea y/o edemas. También
pueden aquejar parestesias, síncope o hipotensión ortostática. Los principales
hallazgos a la exploración física son: hepatomegalia (20%), púrpura (15%),
macroglosia (10%), esplenomegalia (5%) y edemas.
Los síndromes que se asocian a la amiloidosis
primaria son: síndromes nefrótico y del túnel carpiano, insuficiencia cardíaca
congestiva, neuropatía periférica, hipotensión ortostática, malaabsorción y
hepatomegalia masiva (1-4). De hecho, la existencia de cualquiera de estos
síndromes en presencia de un componente monoclonal sérico o urinario es muy
sugestiva de amiloidosis primaria. El ecocardiograma es una técnica muy
rentable en la evaluación de la cardiomiopatía amiloidótica y la biopsia
endomiocárdica es positiva en la práctica totalidad de los casos (6). Los
pacientes que presentan un tabique interventricular igual o superior a 15 mm
tienen una mediana de supervivencia inferior a 6 meses frente a una mediana de
más de dos años para los que tienen un tabique interventricular de grosor igual
o inferior a 12 mm (6). Una sexta parte de los pacientes presentan hipotensión
ortostática (4). Un tercio de los pacientes presentan síndrome nefrótico en el
momento del diagnóstico (5). Alrededor del 15% de los casos tienen neuropatía
periférica. Una cuarta parte de los casos presentan un síndrome del túnel
carpiano (5). En ocasiones, la forma de presentación de la AL consiste en una
hepatomegalia masiva, que simula una cirrosis hepática o sugiere la existencia
de metástasis hepáticas (4,7).
La electroforesis sérica muestra una banda
homogénea de moderada cuantía en la mitad de los casos e hipogammaglobulinemia
en una cuarta parte. Cuando se efectúa una inmunofijación sérica y urinaria se
encuentra componente monoclonal en el 90% de los casos. En el 90% de los casos
existe proteinuria. El uroproteinograma muestra un patrón glomerular con
eliminación predominante de albúmina y en las tres cuartas partes de los casos
la inmunofijación pone de manifiesto la existencia de cadenas ligeras. La
mediana de células plasmáticas en médula ósea en la serie de la Mayo Clinic fue
del 7% (5). En una cuarta parte de los casos existe elevación de la fosfatasa
alcalina. En un 5-10% de los casos existe un déficit de factor X, aunque rara
vez dará lugar a diátesis hemorrágica.
Diagnóstico
El diagnóstico de amiloidosis se basa en la demostración de substancia
amiloide en los tejidos. La posibilidad de una AL debe considerarse en todo
paciente con un componente M sérico o urinario y alguno de los síndromes
propios de la amiloidosis. En el 98% de los pacientes con AL existe componente
M sérico o urinario o bien se puede demostrar una población monoclonal de
células plasmáticas en médula ósea. El procedimiento diagnóstico inicial
consistirá en una punción de la grasa subcutánea, que es positiva en el 80% de
los casos. Si la punción de grasa subcutánea es negativa, el siguiente paso
radica en la práctica de una biopsia rectal, que resulta positiva en el 70% de
los casos. Si tras estas exploraciones no se llega al diagnóstico, se
practicará una biopsia del órgano presumiblemente afecto. Para tipificar la
amiloidosis se efectuará el estudio inmunohistoquímico utilizando anticuerpos
frente a la proteína A, cadenas ligeras kappa y lambda, transtiretina
(prealbúmina ) y beta2-microglobulina. El algoritmo diagnóstico de la AL se
resume en la figura 1.
El diagnóstico diferencial entre amiloidosis primaria y amiloidosis
asociada a mieloma en general no ofrece dificultades (5). Sin embargo, los
limites entre ambas entidades son arbitrarios, ya que ambos procesos son
proliferaciones de células plasmáticas con distinta expresividad clínica.
Siempre que se sospeche una amiloidosis primaria con tinción Rojo Congo
positiva y no se encuentre componente monoclonal en suero y/u orina se debe
desconfiar de este diagnóstico y descartar otras formas de amiloidosis,
particularmente la amiloidosis familiar (1,2). Se debe tener presente que la mediana
de edad en el momento del diagnóstico de la amiloidosis familiar es de 65 años
y que en aproximadamente la mitad de los casos no se encuentran antecedentes
familiares.
Pronóstico
La mediana de supervivencia
de los pacientes con AL es inferior a 2 años y depende fundamentalmente del
síndrome asociado, de tal modo que la mediana de supervivencia es inferior a 6
meses en los pacientes con insuficiencia cardíaca, mientras que es superior a
los 5 años cuando el síndrome asociado es una polineuropatía periférica (5). La
afección cardíaca constituye la causa de muerte en al menos el 50% de los casos
(4,5). La supervivencia actuarial a los 10 años del diagnóstico es ligeramente
inferior al 5% (8).
Tratamiento
El tratamiento de la AL es muy poco satisfactorio (9-11). En una serie de 153 pacientes con AL tratados con melfalán/prednisona, la tasa global de respuestas fue del 20% (11). En esta misma serie, los pacientes con síndrome nefrótico, sin insuficiencia renal ni afección cardíaca, tuvieron una tasa de respuestas del 40%. Se ha demostrado que la colchicina no es eficaz en el tratamiento de la AL (12). Con el empleo de La 4'-iodo-4' -deoxydoxorubicina (I-DOX), derivado antraciclínico con especial afinidad para las fibrillas amiloides, se han referido resultados esperanzadores (13). Por el momento, el tratamiento estándar para los pacientes que no son candidatos a autotrasplante de progenitores hemoperiféricos es la asociación de melfalán y prednisona.
El tratamiento más esperanzador para
pacientes menores de 65 años consiste en dosis elevadas de melfalán seguido de
rescate con progenitores hematopoyéticos de sangre periférica (autotrasplante)
(14). El grupo de Boston ha referido los resultados obtenidos en 103 pacientes
sometidos a dicho procedimiento (15). En el 65% de los pacientes que sobreviven
al año del tratamiento intensivo se alcanza una mejoría objetiva de los órganos
afectos. La probabilidad de alcanzar una respuesta es mayor si el tipo de
cadena ligera es kappa. Sin embargo, en la AL la mortalidad relacionada con el
procedimiento oscila entre el 13 el 43% (14-16). Las causas más frecuentes de mortalidad en el postrasplante inmediato
son: hemorragia digestiva por toxicidad gastrointestinal, insuficiencia
cardíaca y arritmias, rotura de vísceras (intestino, bazo), hipotensión
arteria! refractaria y fallo multiorgánico (15,16). Si bien el tratamiento
intensivo seguido de rescate con progenitores hematopoyéticos mejora la tasa de
respuestas y puede prolongar la supervivencia, dada su elevada mortalidad
resulta imprescindible una cuidadosa selección de los pacientes, limitando la
indicación a los enfermos con afección de solo uno o dos órganos y sin
cardiopatía complicada (15,16). En caso de afección cardíaca grave debe
plantearse la práctica de un trasplante cardíaco y posteriormente, una vez
recuperado de este último procedimiento, proceder al autotrasplante. Los
pacientes con amiloidosis prin1aria posibles candidatos a autotrasplante
deberían ser remitidos a una institución especializada en este tipo de
tratamientos a fin de proceder a una cuidadosa valoración de la indicación. En
caso de que se decida la práctica de un autotrasplante no se debe iniciar el
tratamiento con melfalán y prednisona, ya que el tratamiento alquilante puede
comprometer la obtención de progenitores hemoperiféricos. Alrededor de110% de
los pacientes con AL tienen una afección cardíaca grave predominante con escasa
o nula afectación clínica de otros órganos (1,2). En este caso se debe
considerar la posibilidad de efectuar un trasplante cardíaco, especialmente si
se tienen en cuenta las expectativas creadas por el autotrasplante de
progenitores hematopoyéticos. Las posibilidades terapéuticas en la AL se
resumen en la Tabla 1.
Referencias
1. Falk RH, Comenzo R, Skinner M. The systemic amyloidosis. N Engl J Med
1997; 337: 898-909.
2. Gillmore JD, Hawkins PN, Pepys M Amyloidosis: a review of recent
diagnostic and therapeutic developments. Br J Haematol 1997; 99: 245-256.
3. Kyle RA, Linos A, Beard CM, et al. Incidence and natural history of
primary systemic amyloidosis in Olmstead County, Minnesota, 1950 through 1989.
Blood 1992; 79: 1817-1822.
4. Kyle RA, Gertz MA. Amyloidosis. En: Neoplastic Diseases of the Blood.
2nd Ed. Wiemik PH, Canellos GP, Kyle RA, Schiffer CA, eds. Churchill
Livingstone, Nueva York, 1991; pp: 525-570.
5. Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and
laboratory features in 474 cases. Semin
HematoI 1995; 32: 45-59.
6. Cueto-García L, Reeder
GS, Kyle RA, et al. Echocardiographic findings in systemic amyloidosis: spectrum of cardiac
involvement and relation to survival. J Am Coll Cardiol 1985; 6: 737-743.
7. Gertz MA, Kyle RA. Hepatic amyloidosis (prin1ary -AL-, immunoglobulin
light chain): the natural history in 80 patients. Am J Med 1988; 85: 73-80.
8. Kyle RA, Gertz MA, Greipp PR, et al. Long-term survival (10 year or
more) in 30 patients with primary amyloidosis. Blood 1999; 93: 1062-1066.
9. Kyle RA, Greipp PR. Primary
systemic amyloidosis: comparison of melphalan and prednisone versus placebo.
Blood 1978; 52: 818-827.
10. Kyle RA, Greipp PR, Garton JP, Gertz MA. Primary systemic
amyloidosis: comparison of melphalan/prednisone versus colchicine. Am J Med
1985; 79: 708-716.
11. Gertz MA, Kyle RA, Greipp PR. Response rate and survival in primary
systemic amyloidosis. Blood 1991; 77: 257-262.
12. Kyle RA, Gertz MA, Greipp PR, et al. A trial of three regimens from
primary amyloidosis: colchicine alone, melphalan and prednisone, and melphalan,
prednisone and colchicine. N Engl J Med 1997; 336: 1202-1207.
13. Gianni L, Bellotti V, Gianni AM, Merlini G. New drug therapy of
amyloidosis: resorption of AL type deposits with 4'-iodo- 4'-deoxydoxorubicin.
Blood 1995; 86: 855-861.
14. Comenzo RL, Vosburgh EV, Falk RH, et al. Dose-intensity melphalan
with blood stem cell support for the treatment of AL (amyloid light chain)
amyloidosis: survival and responses in 25 patients. Blood 1998; 91: 3662-3670.
15. Comenzo RL. Hematopoietic cell transplantation for primary amyloidosis:
what have we leamed?. Leuk & Lymph
2000; 37: 245-258.
16. Rives S, Bladé J,
Martínez C, Marín P, Carreras E, Montserrat E. Tratamiento con melfalán a altas
dosis seguido de rescate con progenitores hematopoyéticos en la amiloidosis
primaria. Med Clin(Barc) 2000; 115: 216-220.
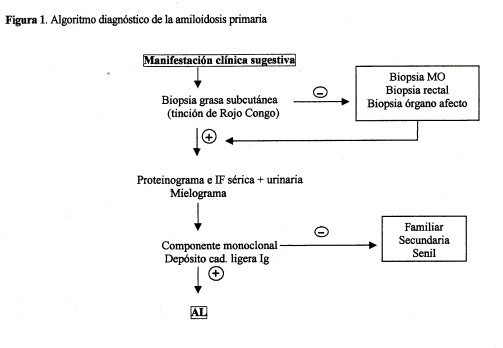
Tabla 1. Opciones terapéuticas en la amiloidosis primaria
|
Melfalán / prednisona(MP) Poliquimioterapia (VBMCP) Dexametasona a dosis altas Colchicina I-DOX |
|
|
Autotrasplante de progenitores hemopoyéticos |
|