
S8. CORRELACION ENTRE EL ASPIRADO MEDULAR, LA BIOPSIA
ÓSEA Y EL INMUNOFENOTIPO EN LINFOMAS DE
CÉLULA PEQUEÑA.
CASO 4
HISTORIA CLÍNICA:
Paciente de 72
años de edad que ingresa en nuestro servicio para estudio de linfocitosis
atípica detectada en un hemograma de control. En los antecedentes destaca
hepatopatía crónica desde hace 15 años. Intervenida de colecistectomía y
coledocotomía por colangitis más colelitiasis con extracción de cálculos y
realización de biopsia hepática, donde se identifican dos espacios portales
mostrándose infiltrado inflamatorio por células redondas. VHB y VHC negativos.
Ecografía abdominal: compatible con hepatopatía crónica. La gamma-GT y las
fosfatasas alcalinas estaban aumentadas. En el hemograma presentaba
leucopenia persistente con leucocitos de 3.8x109/L y 57%
de linfocitos en los últimos 3 años.
Exploración física:
esplenomegalia importante. No se palpan masas tumorales ni adenopatías.
Pruebas complementarias: Hemograma: Hb: 95g/L, VCM: 86.7 fL, leucocitos: 4.2x109/L con presencia de linfocitos atípicos, plaquetas: 100x109/L. En la bioquímica destacaba un aumento de las transaminasas: AST: 1.13 mkat/L (0,005-0,5), ALT: 1,01 mKat/L (0,005-0,63), gamma-GT: 0,86 mKat/L(0,05-0,53), fosfatasas alcalinas: 2,87 mkat/L (0,1- 1,6), uratos: 399 mmol/L (143-371), LDH: 6,6 mKatK/L (0,1-7,6), b2-microglobulina: 6, 8 mg/L (1-2,4) albúmina: 38 g/L, proteínas totales: 67g/L.
La TAC toracoabdominal
detecta esplenomegalia gigante y adenopatías retroperitoneales e ilíacas
derechas.
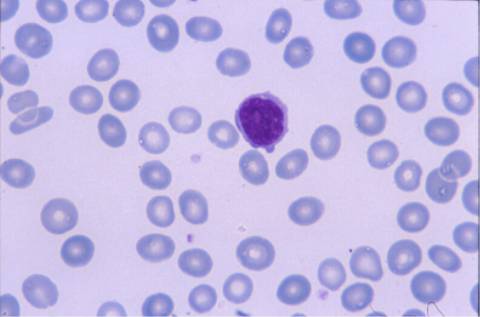
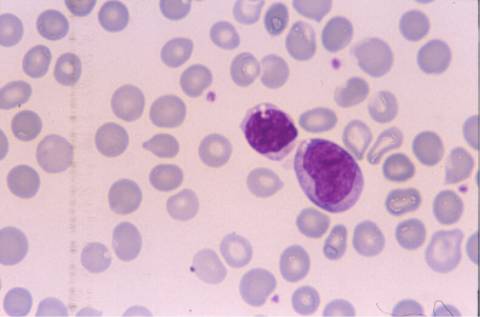
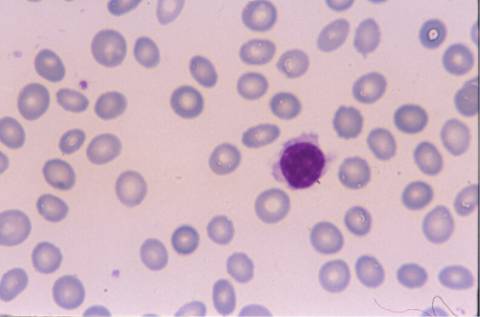
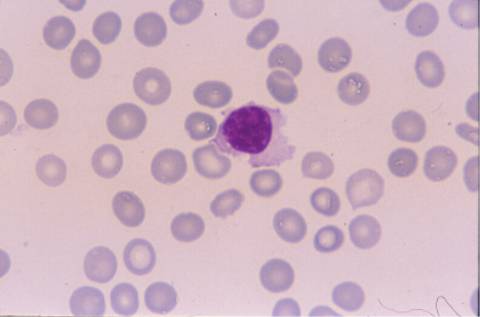
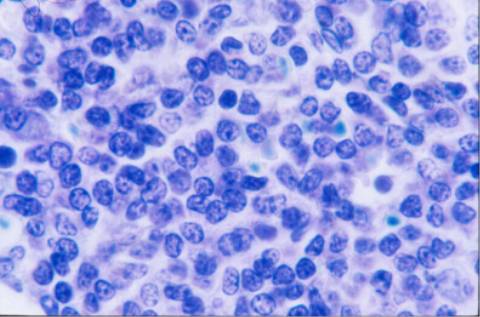
La morfología de los
linfocitos en sangre periférica era muy sugestiva de linfocitos vellosos,
la mayoría de ellos presentaban cromatina madura con un nucléolo pasivo, y
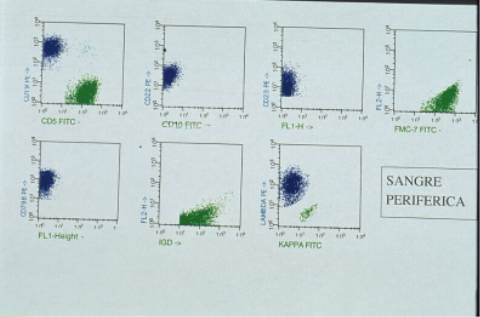
citoplasma visible con prolongaciones vellosas en algunos. El inmunofenotipo
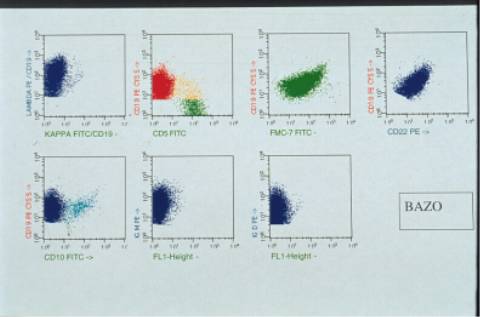
en sangre periférica mostró predominio de linfocitos T (50%) sobre B (31%). La población B CD19
expresaba CD22 (35%), FMC-7 (37% m), CD79b (38% m), CD23 (30% d), lambda (25%
m), IgM +, IgD++. El CD5, CD10, CD38, CD103 y IgS kappa fueron negativos. El
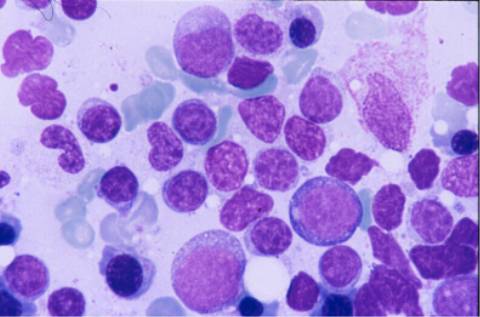
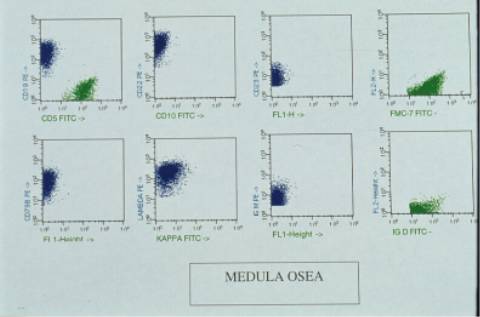
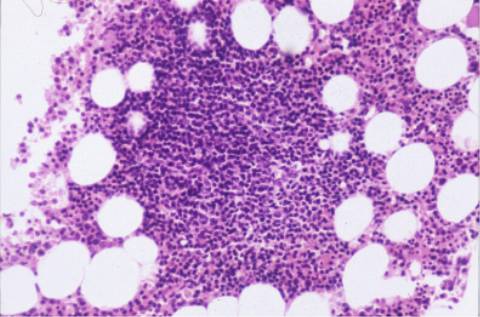
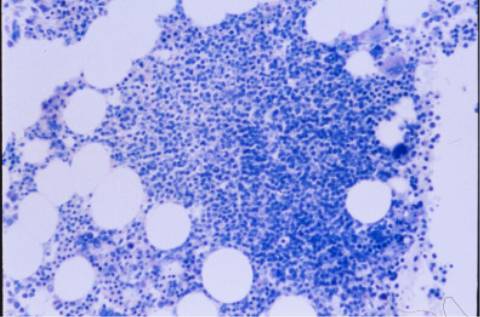
aspirado medular era hipercelular, con
infiltración intersticial del 44% por linfocitos maduros similares a los
de sangre periférica. El inmunofenotipo mostró un predominio de linfocitos B
(49%) sobre los linfocitos T (24%).Restricción de cadenas lambda (43% i), IgM
+, IgD ++ y resto de marcadores B positivos, con CD5 y CD10 negativos como en
sangre periférica.
La morfología junto con el
fenotipo era muy sugestivo de linfoma esplénico de la zona marginal.
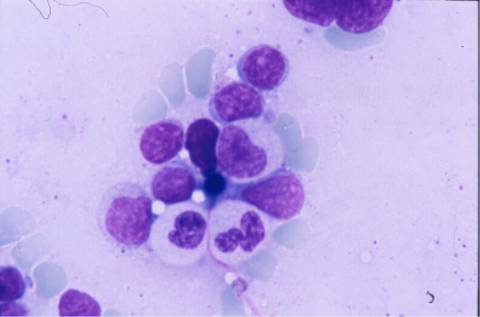
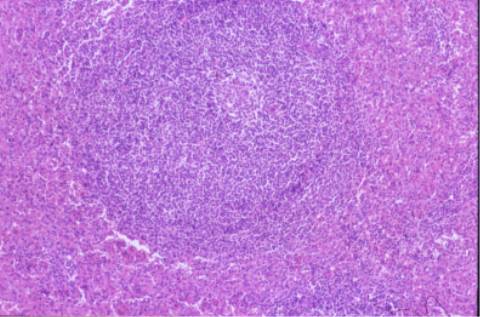
La biopsia ósea mostró una
estructura trabecular normal con presencia de las tres series nobles.
Infiltración intersticial y nodular por linfocitos pequeños, células
plasmáticas y elementos blastoides compatible con síndrome linfoproliferativo
tipo linfoma de células de la zona marginal.
Tratamiento :
Se realizó esplenectomía
extrayéndose un bazo que pesaba 2.100Kg.
La histología esplénica
mostró infiltración por linfoma de células de la zona marginal, más afectación ganglionar
e infiltración sinusoidal hepática. El inmunofenotipo realizado en el bazo fue
superponible a la sangre periférica y médula ósea con predominio casi absoluto
de células B (74%) monoclonales lambda,
CD20: 100%m, CD25 neg, CD11c: 20%, IgM, IgD +++.
Evolución : Hb: 127 g/L,
VCM: 82 fL, plaquetas: 442.0x109/L, leucocitos: 20.0x109/L
con persistencia de un 69% de linfocitos, la mitad de ellos con morfología de
linfocitos vellosos mezclados con linfocitos grandes granulares.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
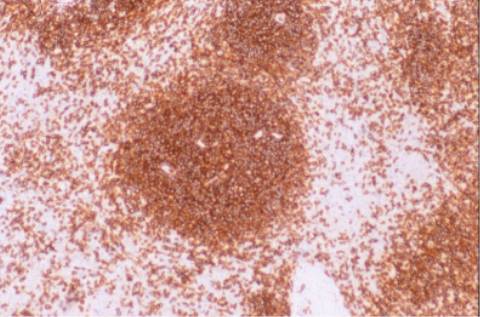
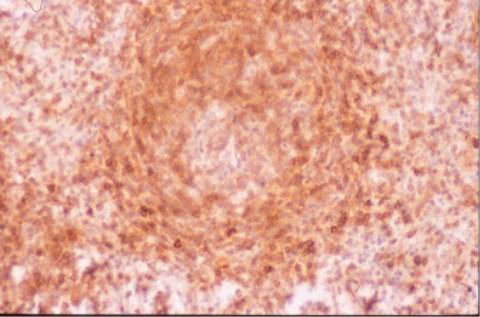
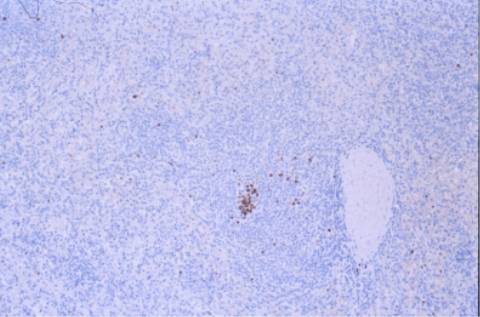
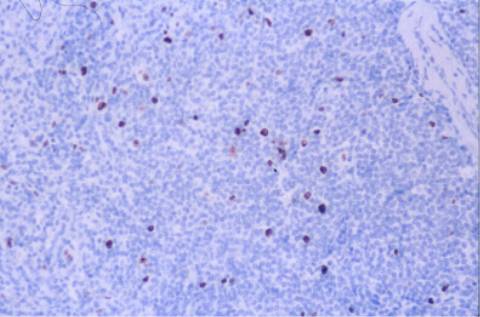
DISCUSION:
El linfoma
esplénico de la zona marginal se incluyó en la clasificación REAL como una entidad provisional ya que no
quedaba claro si las células derivaban de la zona marginal esplénica. En la
clasificación OMS se acepta como entidad, a pesar de que parece que el origen
de este linfoma no está relacionado con las células de la zona marginal
esplénica. Tiene características diferentes de los otros linfomas marginales
(MALT y nodal), como son afectación de la biopsia ósea con infiltración
sinusoidal, expresión en sangre periférica en forma de linfocitos vellosos en
el 50-70% de casos, afectación esplénica en forma de imagen en tres capas en
pulpa blanca y diseminación en pulpa roja, y la mayoría tienen un curso
indolente con buena respuesta a la esplenectomía. Presenta una citología
dimórfica con elementos blastoides y algunos con diferenciación plasmacítica y
un patrón de expresión de Ki-67
bastante característico. No expresa CD5
ni CD10, el CD23 es variable, es IgM e IgD positivo, el bcl-2 es débil y el
bcl-6 negativo. Muchos grupos están trabajando en el origen, y en las bases genéticas
y moleculares de este linfoma. A nivel molecular se observan mutaciones de la
IgV(H) consistentes con que se trata de una célula post-germinal, sin embargo
la ausencia de mutaciones del bcl-6 sugieren que se trate de una célula
pre-folicular. El grupo de F. Solé en una serie de 47 casos de linfoma
esplénico de la zona marginal (SMZL) recientemente publicada, detectan
anomalías de los cromosomas 1, 3, 7, 8, 13, 14 y 20 en la mitad de los casos.
El grupo de Muller-Hermelink estudia 20 casos de SMZL y concluye que este
linfoma en contraste con otros constituye una entidad heterogénea desde el
punto de vista genético, siendo las anomalías mas frecuentes o características
la trisomía 3 y del(7q) y más raras la del(10)(q22-q24) y las delecciones
13q14. El grupo de Catovsky demuestra mediante FISH la presencia de trisomía 3
en algunos linfocitos vellosos circulantes, al igual que se describe en el
bazo. Recientemente el grupo de M.A. Piris ha demostrado que el 40% de estos
linfomas presentan pérdidas alélicas en la región 7q31-q32 lo que constituiría
un marcador genético en esta neoplasia. Este mismo grupo observan que
delecciones del 7q, aumento de blastos y mutación de la p53 constituyen una
forma agresiva de esta entidad. El grupo de E Campo y E.S. Jaffé analizan 36
linfomas de la zona marginal nodales y observan que 6 de los casos presentan
características de linfoma esplénico marginal, son IgD +, con un infiltrado
polimorfo rodeando los centros germinales residuales sin zona del manto y no
cursan con esplenomegalia, ni afectación medular, ni expresión periférica. Los
30 restantes tiene características de linfomas marginales nodales, con células
monocitoides/centrocitoides con infiltración perivascular y perisinusoidal,
centros germinales residuales con zona del manto conservada, y no expresan IgD.
Concluyen que existen linfomas marginales primarios nodales similares al
linfoma esplénico.
En muchas
ocasiones la primera manifestación de este linfoma es la aparición de
linfocitosis absoluta en el hemograma como ocurre en muchas leucemias
linfáticas crónicas. La leucocitosis suele ser moderada (5.0-20.0x109/L)
con una linfocitosis relativa, como en este caso, o absoluta. La morfología
corresponde a linfocitos de cromatina madura que presentan la mayoría un
nucléolo pasivo, el citoplasma está presente, menos amplio que en una
tricoleucemia o tricoleucemia variante, y algunos presentan prolongaciones
vellosas que se disponen a veces en un polo de la célula y que es la
característica que les da el nombre. Pueden acompañarse de linfocitos
transformados o células plasmacíticas. El inmunofenotipo se corresponde con una
célula post-germinal, lo que nos permite separarla en la mayoría de ocasiones
de la LLC; presenta marcadores B de positividad media/intensa, así como la
expresión de IgS (kappa o lambda). El CD10 es negativo, el CD5 es negativo o
positivo débil, el CD23 es variable. Pueden presentar positividad a CD103 como
la tricoleucemia, siendo el CD25 negativo.
En la aspirado
medular el grado de infiltración también es muy variable desde una linfocitosis
intersticial que no llega al 30% hasta grandes agregados que desplazan la
celularidad noble; las características del citoplasma velloso es mas difícil de
valorar y deberemos fijarnos en las
características del núcleo.
En la biopsia
ósea acostumbran a formar agregados
intertrabeculares y cuando la infiltración es intersticial e inferior al 30%
pueden pasar desapercibidos si no se emplea inmunohistoquímica.
Contrario a este caso que hemos presentado, en la práctica de cada día nos encontramos algunos síndromes linfoproliferativos crónicos B, en pacientes sin adenopatías ni esplenomegalia, que presentan en sangre periférica y médula ósea las características descritas en el linfoma esplénico de la zona marginal, tanto morfológicas como fenotípicas. Al no disponer de histología esplénica ni ganglionar será la evolución que determinará o no la esplenectomía, que nos permita confirmar que se trata de un SMZL. La citogenética puede apoyarnos en algunos casos esta sospecha diagnóstica.
Bibliografía
- E. Campo, R. Miquel, L. Krenacs y cols. Primary nodal marginal zone
lymphomas of splenic and MALT type.
Am J Surg Pathol. 23: 59-68, 1999
- Dunn-Walters
DK, Boursier L, Spencer J and Isaacson PG. Analysis of immunoglobulinn genes
in splenic marginal zone lymphoma suggest ongoing mutation. Human Pathol. 29: 585-593,
1998.
- Gruszka-Westwood
AM, Matutes E, Coignet LJ y cols. The incidency of trisomy 3 in splenic
lymphoma with villous lymphocytes: a study by FISH. British J of Haematol,
104: 600-604, 1999.
- NL
Harris, ES Jaffe, J. Diebold y cols. World Health Organization
classification of neoplastic diseasess of the hematopoietic and lymphoid
tissues: report of the Clinical Advisory Committee meeting. J Clin Oncol 17: 3835-3849,
1997.
- M. Mateo, M. Mollejo, R Villuendas y cols. 7q31—32 allelic loss is a frequent
findinng in splenic marginal zone lymphoma. Am J Pathol.,154: 1583-1589,
1999.
- HC
MorseIII, JF Kearney, P. Isaacson y cols. Cells of the marginal zone- origins, function and neoplasia.
Leukemia Research, february: 167-178, 2001.
- Piris MA, Mollejo M; Campo E y cols. A marginal zone pattern may be
found in different varieties of non-Hodking´s lymphoma: the morphology and
immunohistology of splenic involvement by B-cell lymphomas simulating
splenic marginal zone lymphoma. Histopathology, 33: 230-239, 1998.
- F. Solé, M. Salido, B. Espinet y cols. Splenic marginal zone B-cell lymphomas: two
cytogenetic subtypes, one with gain of 3q and the other with loss of 7q.
Haematologica, 86: 71-77, 2001.
- K.
Stein, M. Hummel, P. Korbjuhn y
cols. Monocytoid B cells are distinct from splenc marginal zone cells and
commonly derive from unmutated naive B cells and less frequently from
postgerminal center B cells by policlonal transformation. Blood, 94:
2800-2808, 1999.