
S5
Hallazgos inesperados en Inmunohistoquímica.
Dr. Tomás Álvaro Naranjo / Dr. Joaquín Jaén Martínez
Servicio de Patología. Hospital de Tortosa Virgen de
la Cinta.
C6.
LINFOMA (ENFERMEDAD) DE HODGKIN
ÍNDICE
DESCRIPCIÓN DEL
CASO
Hombre
de 38 años de edad con antecedentes de amigdalectomía a los dos años.
Consulta
en la actualidad por presentar adenomegalias múltiples axilares bilaterales de
hasta 5 cm de tamaño. La exploración muestra también una adenopatía inguinal
derecha de 1 cm de diámetro.
Analítica:
Leucocitos 14500, Hb 12´4, plaquetas 355000.
Masa
bulky mediastínica. La ecografía abdominal muestra una leve hepatomegalia del
lóbulo izquierdo y en el bazo se observan múltiples nódulos hipoecogénicos de 5
a 15 mm. Adenopatías retroperitoneales paraórticas y celíacas de 25 a 30 mm.
Evolución:
Enfermedad de Hodgkin, Esclerosis nodular, Estadio III A. Tratamiento según
esquema híbrido C-MOPP/ABV.
Resulta
refractario al tratamiento, y a los 12 meses del diagnóstico inicial se inicia
tratamiento de rescate (mini-BEAM) y se plantea auto-TMO. En el curso del
tratamiento síndrome febril en el contexto de neutropenia postquimioterapia,
del que se recupera tras tratamiento con ceftazidima y amikacina junto con
G-CSF. Remisión parcial.
Al
segundo año tras el diagnóstico, se realiza auto-TMO, sin respuesta,
apareciendo nueva recidiva con infiltración pleural y derrame pleural. Se
inicia tratamiento radioterápico.
Vivo
con enfermedad a los tres años del diagnóstico.
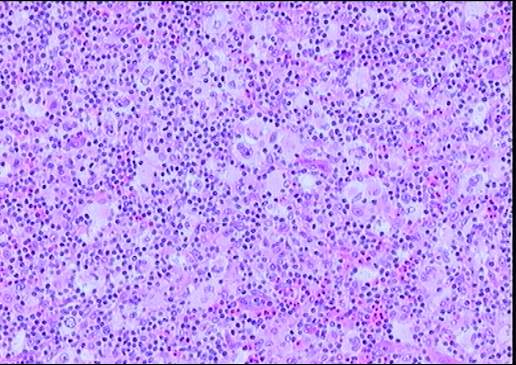
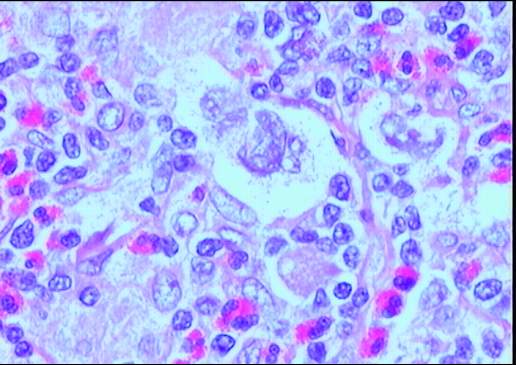
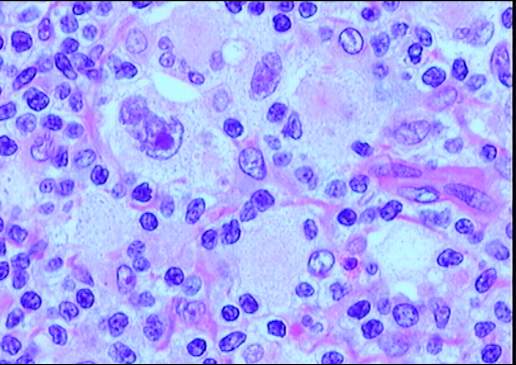
Descripción morfologica e inmunofenotipica:
Ganglio linfático con estructura general alterada
incluyendo engrosamiento y fibrosis capsular, tractos escleróticos y una
desorganización arquitectural del parénquima a expensas de numerosas células
grandes con intenso pleomorfismo, uni o
multinucleadas, con nucléolos eosinófilos prominentes y citoplasma amplio y
claro con frecuente artefacto o retracción periférica que produce imagen típica
de célula lacunar. Presencia de células de Reed-Sternberg. Existe una
celularidad acompañante de fondo constituida por numerosos histiocitos,
polinucleares eosinófilos, células plasmáticas y linfocitos. Frecuentes y
amplias zonas de necrosis.
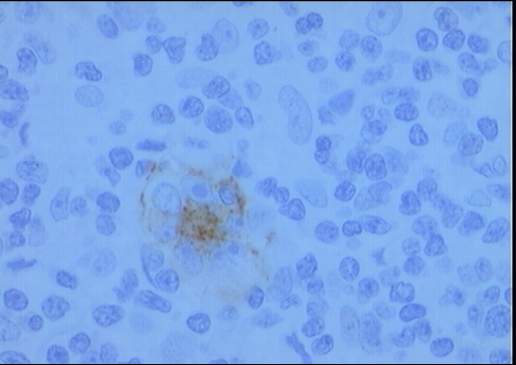
El estudio inmunohistoquímico muestra un perfil
sobre la celularidad atípica CD45 negativo y CD15 positivo con ocasionales
células atípicas positivas para CD20 (10%). LMP-1 y CD30 son negativos, o con
tinción muy débil y ocasional para el último. La celularidad linfoide
acompañante corresponde de forma mayoritaria a linfocitos T (CD3).
|
H.E. |
H.E |
H.E. |
CD15 |
CD15 |
|
CD30 |
CD30 |
CD45 |
CD45 |
CD20 |
|
CD20 |
CD20 |
CD20 |
CD3 |
P53 |
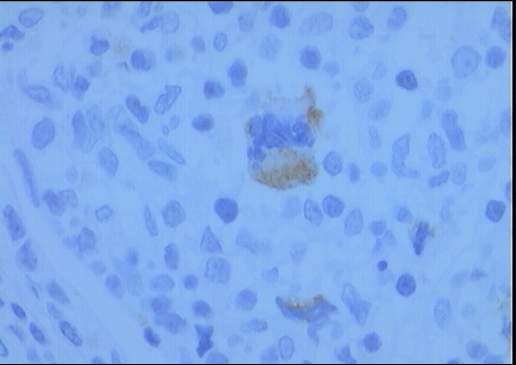
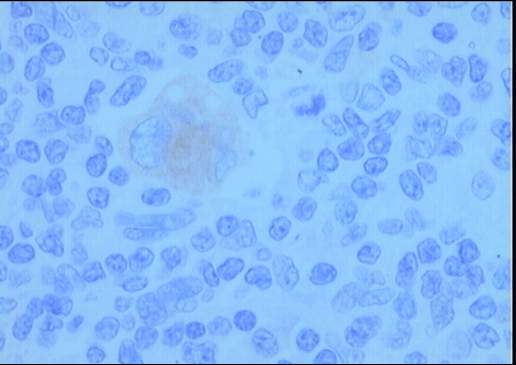
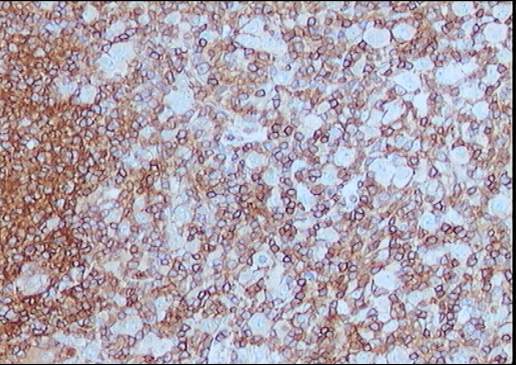
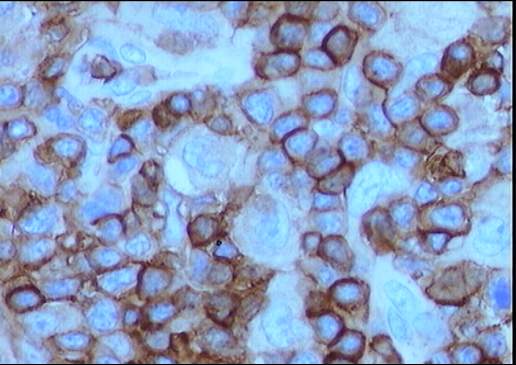
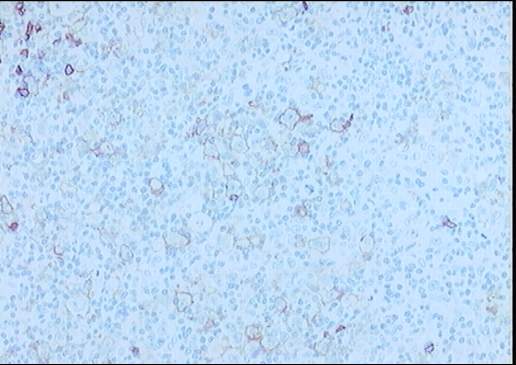
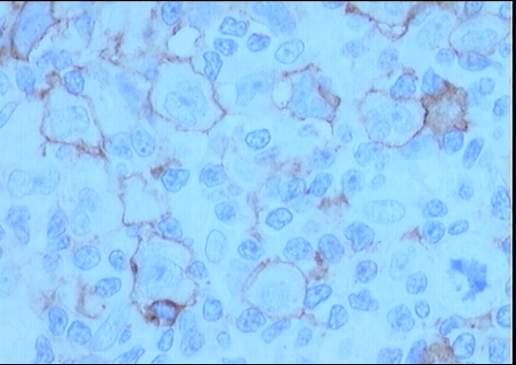
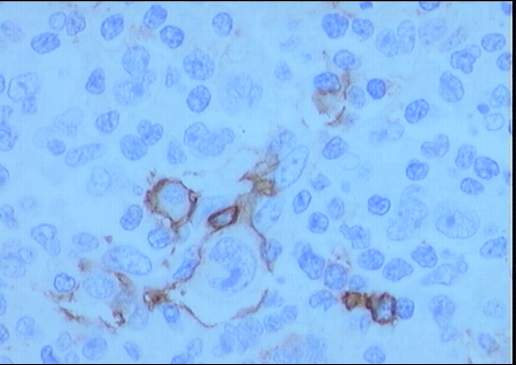
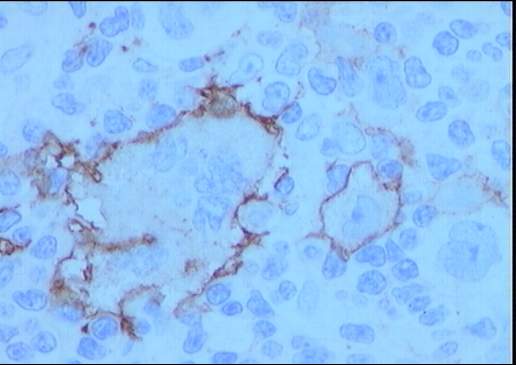
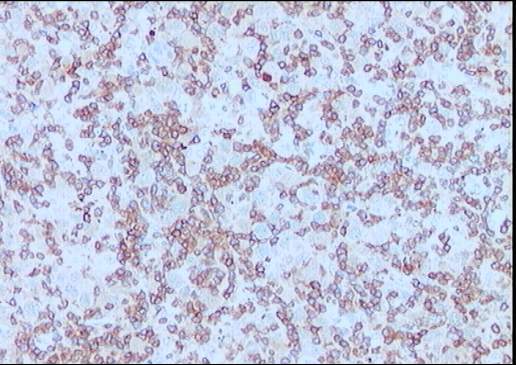
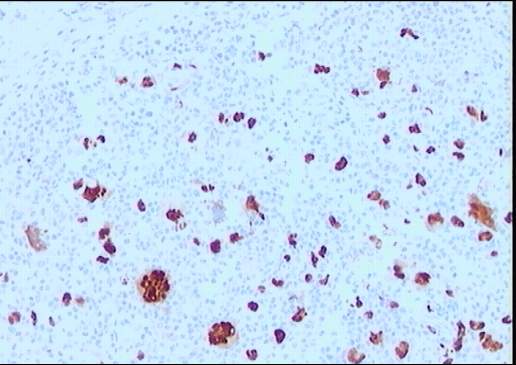
Diagnóstico:
GANGLIO LINFÁTICO (REGIÓN
AXILAR DERECHA):
- ENFERMEDAD DE HODGKIN, ESCLEROSIS NODULAR.
DISCUSIÓN
Introducción
Desde 1832 en que fue descrita por primera vez
(Hodgkin 1832), múltiples teorías han caracterizado la enfermedad de Hodgkin
(EH) como proceso reactivo / tumoral, así como su elemento celular principal. A
día de hoy, sólo respecto de la naturaleza neoplásica de la enfermedad parece
existir consenso, hablándose ahora ya de linfoma y no de enfermedad de Hodgkin,
pero continuando el debate entorno al origen de la célula proliferante que
caracteriza el proceso. Además, y pese a ser una enfermedad bien conocida, sigue
planteando numerosas situaciones de auténtica dificultad en el terreno del
diagnóstico diferencial, tanto en lo que se refiere a su reconocimiento, con
numerosas variedades y apariencias morfológicas; como a su inmunofenotipo, con
frecuentes excepciones al perfil típico; como desde el punto de vista
conceptual, con aspectos cercanos a linfomas no Hodgkin B, linfoma anaplásico y
otros.
Para
poner un ejemplo reciente, Cazals-Hatem D, et al, 2001, han comunicado 77
pacientes con enfermedad de Hodgkin que fueron diagnosticados y tratados
erróneamente como linfoma no Hodgkin. Los tres hematopatólogos que hicieron la
revisión cambiaron el diagnóstico de 46 casos basándose en datos morfológicos,
que habían sido interpretados inicialmente como linfoma anaplásico-Hodgkin like
(ALCL-HL), linfoma anaplásico, linfoma periférico de células T o linfomas B.
Los otros 31 casos, que habían sido considerados por dicho panel como ALCL-HL o
linfoma periférico de células T, sólo fueron finalmente identificados como EH
tras el estudio inmunofenotípico. Las consecuencias clínicas del tratamiento
como linfoma no Hodgkin de los casos de EH fue una disminución del tiempo de
supervivencia, probablemente debido a un mayor número de recaídas.
Uno
de los avances recientes que ha contribuido a aclarar parte de esta confusión
es el análisis genético que permite reconocer en el ALCL la t(2;5) o alguna de
sus variantes, con sobreexpresión de la proteína ALK, que no es encontrada en
la EH. Así pues, el grupo de linfomas llamados inicialmente ALCL-HL han dejado
de ser considerados como ALCL y son considerados en la actualidad EH.
Con
respecto a la confusión con linfomas B, al menos dos son las situaciones que
nos podemos encontrar. La primera es el diagnóstico diferencial entre
paragranuloma y linfoma B rico en células T, con CD15-, CD30-, EMA+ y CD20+ en
ambos casos. La otra es el hallazgo de células CD20+ en la EH, como ilustra el
caso que presentamos en este seminario, y que se encontró en 7 de los casos
comunicados por Cazals-Hatem D, et al, 2001, motivo por el que fueron
erróneamente diagnosticados como linfoma no Hodgkin. Cabe añadir aquí que 9 de
sus casos mostraron positividad para CD3 en células de Hodgkin y R-S, aunque
con patrón citoplásmico y nunca de membrana, razón por la que habían sido
diagnosticados de linfoma T o ALCL. Finalmente se objetivó positividad para EMA
en 7 casos, para CD45 en 10, CD43 en dos y TIA1 en 8. Todos fueron negativos
para ALK, un 31% para CD15 y 5% fueron negativos para CD30.
Clasificación y célula
de origen
Tal y como ha ocurrido con otros síndromes
linfoproliferativos, se mantiene la idea de que la identificación de la célula
de origen de la EH permitiría una conceptualización correcta de la enfermedad,
con una mejor clasificación de la misma y el nacimiento de nuevos abordajes
diagnósticos y terapéuticos.
Determinada por la presencia de células de Hodgkin y
de Reed-Sternberg diagnósticas, sobre un ambiente celular reactivo
característico, la EH ha gozado de amplio consenso taxonómico, con escasas variaciones
a lo largo de los años. Las recientes clasificaciones REAL (Harris 1994,
Morente 1995) y OMS (Jaffe 1997, Álvaro 1997) identifican cinco categorías de la enfermedad (Tabla 1) dentro de dos
grupos principales, enfermedad de Hodgkin Clásica (EHc) y enfermedad de Hodgkin
predominio linfocítico (PLN) con marcadas diferencias respecto a la morfología
e inmunofenotipo de la célula tumoral así como respecto de la composición del
ambiente celular no neoplásico
acompañante.
|
Tabla 1 |
|
|
|
REAL |
OMS |
|
|
Lymphocite predominance Lymphocite-rich classic Nodular
sclerosis Mixed cellularity Lymphocytic depletion |
Nodular Lymphocite predominance Lymphocite-rich classic Nodular
sclerosis (grades I – II) Mixed cellularity Lymphocytic depletion |
|
En
los últimos años el análisis de los genes de las células de R-S mediante
amplificación por PCR en células obtenidas por micro manipulación han supuesto
un avance considerable en la comprensión conceptual de la EH. Las evidencias
apuntan hacia un origen sobre células B post-germinales de las células de R-S y
un origen sobre células B del centro germinal para las células L&H. Pero
también existen datos que sugieren que al menos en una pequeña proporción de
casos las células de R-S derivan de linfocitos T, especialmente aquellos que
contienen moléculas citotóxicas.
También
el papel del virus de Epstein-Barr ha sido destacado en la EH, a través del
efecto de LMP-1 sobre otras proteínas como TNF y el factor de transcripción NF-kB y debido al déficit de respuesta de las células-T citotóxicas a
LMP-1.
Además
las manifestaciones clínicas de la enfermedad, incluyendo los síntomas B y la inmunodepresión derivan del efecto de
las citocinas liberadas por el tumor, así como el resto del micro ambiente
celular y tejido fibroso característicos de la enfermedad (Kadin ME, 2001).
¿Sólo dos formas de enfermedad de Hodgkin?
El PLN constituye el 5% del total de casos de EH con
predominio en varones entre 25 y 45 años, que suelen presentarse en estadios
clínicos precoces con respuesta completa al tratamiento en el 90% de los casos.
La célula tumoral, L&H, evidencia rasgos inmunohistoquímicos diferenciales
respecto a la clásica célula de Reed-Sternberg con inmunorreactividad frente a
CD45/45RB, CD20, cadena J de inmunoglobulinas y ausencia de expresión de CD30 y
CD15. La identificación de mutaciones somáticas en los genes de las regiones
variables de las inmunoglobulinas establece el origen en células B de centro
germinal.
La EHc muestra una presentación bimodal con un pico de incidencia entre los 15-35
años sin predominio por sexos, buen pronóstico y predominio del tipo esclerosis
nodular. El segundo pico, por encima de los 50 años con predominio en varones,
mayor incidencia del tipo celularidad mixta y curso clínico más agresivo. La
población celular tumoral (célula de Reed-Sternberg y sus variantes) no supera
en ocasiones el 1% del total, con inmunofenotipo clásico CD30+, CD15+, CD45-
(Vimentina +, Fascina +, CD40+, CD20 -, CD3 -, EMA -, ALK-). Merece la pena
destacar que han sido comunicadas excepciones en la expresión esperada prácticamente
para todos y cada uno de los marcadores mencionados, a veces incluso con
implicaciones de tipo pronóstico, como ocurre en la negatividad de CD15, que
supone un factor pronóstico negativo independiente para recidivas y
supervivencia (von Wasielewski R, et al. 1997).
La diferenciación entre PLN y EHc ha encontrado
recientemente nuevo eco. Anagnostopoulos I. et al. 2000, revisaron 388
pacientes con el diagnóstico de PLN. Tras la revisión sólo el 56% de los casos
fueron confirmados, debido fundamentalmente al reconocimiento de una nueva
variante de EHc, también con patrón nodular y muy rica en linfocitos,
denominada enfermedad de Hodgkin clásica nodular rica en linfocitos. Con un
inmunofenotipo clásico, CD30 y/o CD15+, casi un tercio de los casos muestra
positividad para CD20, 13% para CD79a y un 6% para EMA. Clínicamente estos
pacientes presentaron unas características diferentes tanto del PLN como del
resto de formas de EHc.
El PLN
muestra un inmunofenotipo característico distinto del resto de tipos de EHc, lo
que ha servido para sugerir desde hace tiempo que esta enfermedad compuesta por
células B y derivada de células del centro germinal, debería ser clasificada
como un Linfoma no Hodgkin. Y, en efecto, las células L&H expresan
consistentemente CD45 y marcadores de
línea B, como CD20, y frecuentemente EMA. Más difícil es la demostración de
restricción de cadenas ligeras de las Ig, al menos por métodos
inmunohistoquímicos, aunque puede ser demostrada por hibridación in situ
(Stoler MH, et al, 1995). Además, y a diferencia de en la EHc, las células
L&H no expresan vimentina, CD15 y casi nunca CD30 (Rudiger T, et al, 1998).
Son características las rosetas de células CD57+ alrededor de las células
L&H, aunque ni se ven en todos los casos ni cuando se ven son enteramente
específicas de esta enfermedad.
La EHc rica en linfocitos ha sido incorporada en la
clasificación de la OMS como un tumor difuso, con pocas células R-S sobre un
fondo de linfocitos con pocos eosinófilos y células plasmáticas. El diagnóstico
diferencial que se plantea es con la rara forma difusa del predominio
linfocítico y, en menor medida, con las fases celulares de la esclerosis
nodular o la celularidad mixta. Pero en contraste con el predominio
linfocítico, aquí las células tumorales muestran el inmunofenotipo
característico R-S que ya ha sido mencionado más arriba, aunque también pueden
expresar CD20. Además existe una variante folicular o nodular de esta enfermedad
con las mismas características (Ahston-Key M, et al, 1995).
Los estudios inmunofenotípicos juegan un papel
decisivo en la distinción entre predominio linfocítico y EHc, variando entre el
12-24% las interpretaciones morfológicas (von Wasielewski R, et al. 1997), con
el consiguiente impacto sobre el curso clínico. Otro estudio más reciente ha
demostrado el valor del inmunofenotipo en la clasificación del predominio
linfocítico y la EHc rica en linfocitos (Diehl V, et al, 1999). De 426 casos
diagnosticados como predominio linfocítico, sólo el 51% fue confirmado tras la
revisión histológica y el estudio inmunohistoquímico. El resto correspondieron
115 casos (27%) a EHc rica en linfocitos, y el resto a una mezcla de EH, LNH y
lesiones reactivas.
¿Puede
considerarse a las células de Hodgkin y R-S de la EHc como células B?
Las
células de Hodgkin y de R-S representan una población clonal derivada la
mayoría de las veces de células B predestinadas a morir, pero que escapan del
proceso apoptótico a través de algún mecanismo transformante. Las evidencias de
un origen B son múltiples.
Existen
numerosas observaciones de casos de EH que expresan antígenos normalmente
asociados a células B (Korkolopoulou P, et al. 1994; Schmid C, et al, 1991;
Siebert JD, et al, 1995; Zukerberg LR, et al, 1991; Vasef MA, et al, 1997). Al
menos CD20, CD79a, CD19, CD138 y PCA-1 han sido mostrados en un número variable
de casos (Watanabe K, et al, 2000).
El factor de transcripción específico de células B
(BSAP) está codificado por el gen Pax-5, expresado por todas las células B
excepto por células plasmáticas terminalmente diferenciadas. Foss et al (1999),
identifican expresión de BSAP mediante técnicas inmunohistoquímicas y de
hibridación in situ en el 80% de casos de EHc y en todos los PLN, confirmando
el origen en células B de estos procesos. Considerando el papel de BSAP en
diferentes funciones, incluyendo el incremento de expresión de antígenos de
células B como CD19 y CD20, la ausencia de expresión de estos marcadores por
parte de las células tumorales es atribuida a la débil-moderada expresión de
BSAP detectada.
La proteína BCL6 es un represor transcripcional en
dedos de zinc codificada por el protooncogén BCL6. En condiciones fisiológicas,
es expresada por células B del centro germinal pero no por precursores
inmaduros de células B ni células plasmáticas, siendo necesaria para la
formación y función de los centros germinales( Cattoretti 1995, Dent 1997).
Falini et al (1996) observan la constante expresión
de BCL-6 por parte de las células L&H (100%) así como sobre la celularidad
reactiva acompañante con ausencia de expresión de CD40 ligando. Todo ello
vendría a corroborar un distintivo origen celular en los casos de PLN a partir
de células B centro germinales, frente a EH clásica, en la que la expresión
sólo se identifica en el 30 % de los casos, con un fenotipo BCL-6 - / CD40L+ de
los linfocitos T CD3+ / CD4+ acompañantes.
Por su parte, Carbone et al. (1998) establecen la
existencia de dos categorías fenotípicas de la enfermedad de Hodgkin, sobre la
base de la expresión de BCL6 y Syn-1. Syn-1 es un proteoglicano implicado en la
interacción célula-matriz extracelular expresado por células B post-centro germinal,
incluyendo inmunoblastos y células plasmáticas. Los resultados muestran la
existencia de dos variantes de expresión. Por un lado, BCL-6+ / syn-1 –
establecería un origen celular a partir de célula B de centro germinal,
habiéndose evidenciado la expresión en el 100% de casos de PLN. En contraste,
Bcl-6- / syn-1+, indicativo de origen en célula B postgerminal, es expresado
por células RS en la mayoría de casos de enfermedad de Hodgkin clásica.
Diversos trabajos concluyen en la misma línea cuando
se tipifica la enfermedad de Hodgkin respecto a la expresión de BCL-6, no
obteniéndose expresión de otros marcadores de células B centro foliculares como
CD10 (Dogan 2000).
El origen postgerminal de la célula de RS,
identificado mediante la determinación de la expresión de BCL-6- / syn-1 +
(Carbone 1998) en la mayoría de casos de E. H. clásica es establecido,
asimismo, en los casos asociados a infección VIH (Carbone 1999), así como
mediante el estudio de la expresión de MUM-1 (Carbone 2001). MUM1 / IRF 4 es un
factor de transcripción perteneciente a la familia del factor regulador del
interferón, con especificidad linfocitaria, identificado por su participación
en la traslocación t(6;14)(p25;q32) del mieloma múltiple (Lida 1997) y
considerado marcador del paso final en la diferenciación de células B intra-CG
y de la maduración hacia células plasmáticas (Falini 2000). Se observa una
consistente expresión de BCL-6 - / MUM-1 + / Syn-1 +, indicativo de un origen
en célula B post-CG en el 100% de los casos estudiados, representativos todos
ellos del espectro patológico de EH Clásica.
En el timo las células B medulares han sido
apuntadas como el origen de las células de Hodgkin y RS, especialmente en
función de la positividad para CD20 observado en ellas (Krugmann J, et al,
1999).
¿ Estirpe T ?
Seguramente
una de las principales dificultades en el terreno del diagnóstico diferencial
de la EH se plantea con ALCL o linfoma T. Ya se ha mencionado que puede
encontrarse positividad para CD3 en células de Hodgkin en más del 10% de los
casos, aunque de forma globular intra citoplásmica y no de membrana. Este
hallazgo, reconocido desde hace tiempo por Cibull ML, et al, 1989, dio pie a
pensar que al menos algunos casos de EH podían originarse sobre linfocitos T
activados. En la práctica, la investigación de reordenamiento para TCR puede
ayudar a realizar el diagnóstico diferencial entre EH y linfoma T con presencia
de células de tipo RS-like (Quintanilla-Martinez L, et al, 1999). La mayoría de
estos casos de EH muestra reordenamiento clonal para el gen de las Ig, y no hay
reordenamiento para TCR a pesar de la expresión citoplásmica de CD3. Pero el
15% de EH con expresión de CD3, es decir, entre 1-2% de todos los casos de EHc,
muestran reordenamiento para TCR gamma (Seitz, V, et al, 2000), mostrando pues
una derivación T.
El
mismo grupo (Seitz, V, et al, 2001) ha comunicado que todos los casos de EHc
derivados de células B presentan mutaciones de BCL-6, en contraste con aquellos
derivados de células T, que no lo hacen.
Finalmente,
el hallazgo de granzima B, perforina y TIA-1 no constituye una rareza en
células de Hodgkin y R-S (Oudejans JJ, et al, 1996), lo que ha servido para
hipotetizar un posible origen a partir de células T citotóxicas activadas o incluso
de células NK. Por cierto que también parece posible que células de R-S
derivadas de células B maduras expresen de forma aberrante moléculas
citotóxicas T (Muschen M, et al, 2000). La posibilidad de que las células de
Hodgkin y R-S que coexpresen diferentes marcadores representen fusiones
celulares ha sido, de momento, descartada (Kuppers R, et al, 2001).
Células
dendríticas foliculares
Existen
evidencias que parecen sugerir que bajo el término de enfermedad o linfoma de
Hodgkin se incluyen un grupo de enfermedades diversas, y no sólo en cuanto a
presentación morfológica o curso clínico, sino también en lo que se refiere a
la célula que da origen al tumor.
Profundizando
en esa idea, se han comunicado una serie de casos de EH con morfología e
inmunofenotipo característicos (CD30, CD15), que presentan marcadores de
células dendríticas foliculares como CD21 (Delsol G, et al, 1993), y además
también DRC1, CD35, R4/23 y Ki-M4p (Nakamura S, et al, 1999). Ninguno de estos
casos presentaba marcadores de células B y, en ellos, el estudio del genotipo
mostró configuración en línea germinal tanto para el gen TCR como para el de
cadenas pesadas de las Ig.
Células dendríticas interdigitantes:
También
se ha sugerido una posible relación de estas células con EH. Aquí los
argumentos empleados son una morfología de las células de Hodgkin y R-S similar
a las células dendríticas interdigitantes, una estrecha interacción con las
células T del entorno inmediato, que ambos tipos celulares presentan en común,
y la expresión de fascina (Pinkus GS, et al. 1997).
Inmunobiología y carga tumoral
La EH es un tumor de células de Hodgkin y R-S
secretoras de citocinas, capaces de modular la sintomatología clínica y la
apariencia histológica de la enfermedad (Kadin ME, 2001). Que se sepa, la
eosinofilia puede ser atribuida a la secreción de IL-5 y eotaxina, la
hiperplasia mieloide acompañante a la secreción de GCSF, la esclerosis nodular
a TGF-beta1 y factor de crecimiento fibroblástico, la supresión inmune también
a TGF-beta1 y los síntomas B a IL-6.
El paciente presentado en este seminario tenía masa
bulky en la presentación. Existen evidencias de la significación pronóstica de
la carga tumoral en enfermedad de Hodgkin y renovados esfuerzos por definirla
con exactitud (Gobbi PG, et al, 2001). Posiblemente los niveles de IL-2R y de
CD30 son asimismo marcadores de carga tumoral (Kadin ME, 2001).
Quedan por contestar todavía algunas cuestiones que
plantea la EH en la actualidad, que se presenta como un grupo de enfermedades
con orígenes diversos, con implicaciones etiológicas desconocidas más allá del
papel del VEB, con apariencias morfológicas y acompañamiento reactivo muy
diverso y con inmunofenotipo variable, como es el caso que se presenta en este
seminario, con expresión de CD20 y en cambio ausencia de CD30. ¿Poseen estos
hallazgos significación en la refractariedad al tratamiento de este paciente?
BIBLIOGRAFÍA
Álvaro
T , García B. Clasificación de la
Organización Mundial de la Salud (OMS) de los tumores del tejido hematopoyético
y linfoide. En: ”4 Curso de Hematopatología”, Ed.: Tomás Álvaro Naranjo,
Llorenç Font Ferré, Ramón Bosch Príncep, M. Teresa Salvadó Usach, Josep Gumá
Padró.
Ahston-Key M, Thorpe PA, Allen JP, et al.
Follicular Hodgkin´s disease. Am J Surg Pathol 1995; 19: 1294-1299.
Anagnostopoulos
I, Hansmann ML, Franssila K, Harris M, Harris NL, Jaffe ES, Han J, van Krieken
JM, Poppema S, Marafioti T, Franklin J, Sextro M, Diehl V, Stein H. European
Task Force on Lymphoma project on lymphocyte predominance Hodgkin disease:
histologic and immunohistologic analysis of submitted cases reveals 2 types of
Hodgkin disease with a nodular growth pattern and abundant lymphocytes. Blood.
2000 Sep 1;96(5):1889-99.
Baron
BW, Nucifora G, McCabe N, Espinosa R, Le Beau MM, McKeithan TW. Identification of the gene associated with the recurring chromosomal
translocations t(3;14)(q27;q32) and t(3;22)(q27;q11) in B-cell lymphomas. Proc Natl Acad Sci USA 1993;
90: 5262.
Carbone A, Gloghini A,Gaidano G, Franceschi S,
Capello D, Drexler HG, Falini B, Dalla-Favera R. Expression status of BCL-6 and
syndecan-1 identifies distinct histogenetic subtypes of Hodgkin’s disease.
Blood 1998 ; 92 : 2220-2228.
Carbone A, Gloghini A, Larocca LM, Antinori A,
Falini B, Tirelli U, Dalla-Favera R, Gaidano G. Human immunodeficiency
virus-associated Hodgkin's disease derives from post-germinal center B cells.
Blood 1999 Apr 1;93(7):2319-26 .
Carbone A, Gloghini A, Larocca LM, Capello D,
Pierconti F, Canzonieri V, Tirelli U, Dalla-Favera R, Gaidano G. Expression
profile of MUM1/IRF4, BCL-6, and CD138/syndecan-1 defines novel histogenetic
subsets of human immunodeficiency virus-related lymphomas. Blood 2001 Feb
1;97(3):744-51
Cattoretti G, Chang C-C, Cechova K, Zhang J, Ye
BH, Falini B, Louie DC, Offit K, Chaganti RSK, Dalla-Favera R. BCL-6 protein in
expressed in germinal-center B cells. Blood 1995 ; 86: 45.
Cazals-Hatem D, Andre M, Mounier N, Copin MC, Divine M, Berger F,
Bosly A, Kerneis Y, Briere J, Quesnel B, Diebold J, Gaulard P. Pathologic and
clinical features of 77 Hodgkin's lymphoma patients treated in a lymphoma
protocol (LNH87): a GELA study. Am J Surg Pathol. 2001 Mar; 25(3): 297-306.
Cibull ML,
Stein H, Gatter KC, Mason DY. The expression of the CD3 antigen in Hodgkin's
disease. Histopathology. 1989 Dec;15(6):599-605.
Delsol G, Meggetto F, Brousset P, et al,
Relation of follicular dendritic reticulum cells to Reed-Sternberg cells of
Hodgkin´s disease with emphasis on the expression of CD21 antigen. Am J
Pathol 1993; 142: 1729-1738.
Dent AL, Shaffer AL, Yu X, Allman D, Staudt LM.
Control of inflammation, cytokine expresion, and germinal center formation by
BCL-6. Science 1997; 276: 589.
Diehl V, Sextro M, Franklin J, Hansmann ML,
Harris N, Jaffe E, Poppema S, Harris M, Franssila K, van Krieken J, Marafioti
T, Anagnostopoulos I, Stein H. Clinical presentation, course, and prognostic
factors in lymphocyte-predominant Hodgkin's disease and lymphocyte-rich
classical Hodgkin's disease: report from the European Task Force on Lymphoma
Project on Lymphocyte-Predominant Hodgkin's Disease. J Clin Oncol. 1999 Mar; 17(3): 776-83.
Dogan A, Bagdi E, Munson P, Isaacson PG. CD10 and BCL-6 expression in paraffin sections of normal lymphoid tissue
and B-cell lymphomas. Am J Surg Pathol 2000; 24: 846-52.
Falini
B, Bigerna B, Fizzotti M, et al. Distinctive expression
pattern of the BCL-6 protein in nodular lymphocyte predominance Hodghin’s
disease. Blood 1996; 87: 465-71.
Falini
B, Fizzotti M, Pucciarini A, et al. A monoclonal antibody
(MUM1p) detects expression of the MUM1/IRF4 protein in a subset of germinal
center B cells, plasma cells and activated T cells. Blood 2000; 95: 2084-2092.
Foss HD, Reusch R, Demel G, Lenz G, Anagnostopoulos
I, Hummel M, Stein H. Frequent expression of the B-cell-specific activator
protein in Reed-Sternberg cells of classical Hodgkin’s disease provides further
evidence for its B-cell origin. Blood 1999; 9: 3108-3113.
Gobbi PG, Ghirardelli ML, Solcia M, Di
Giulio G, Merli F, Tavecchia L, Berte R, Davini O, Levis A, Broglia C, Maffe
GC, Ilariucci F, Dore R, Ascari E. Image-aided estimate of tumor burden in
Hodgkin's disease: evidence of its primary prognostic importance. J Clin Oncol.
2001 Mar 1; 19(5): 1388-94.
Harris NL, Jaffe ES, Stein H, et al. A revised
European-American classification of lymphoid neoplasms: a proposal from the
International Lymphoma Study Group. Blood 1994; 84: 1361-1392.
Jaffe
ES. Introduction to the WHO classification. Am J Surg
Pathol 1997; 21: 114-121.
Kadin ME. Hodghin’s disease: cell of origin,
immunobiology, and pathogenesis. En: Knowles, DM. Neoplastic Hematopathology,
sedond edition. Lippincott Williams&Wilkins, Philadelphia 2001.
Kerckaert J-P, Deweindt C, Tilly H, Quief S,
Lecoq G, Bastrad C. LAZ3, a novel zinc-finger encoding gene, is disrupted by
recurring chromosome 3q27 translocations in human lymphomas. Nat Genet 1993; 5:
66.
Korkolopoulou P, Cordell J, Jones M, Kaklamanis
L, Tsenga A, Gatter KC, Mason DY. The expression of the B-cell marker mb-1
(CD79a) in Hodgkin's disease. Histopathology. 1994 Jun;24(6):511-5.
Krugmann J, Feichtinger H, Greil R, Fend F.
Thymic Hodgkin´s disease -- a histological and immunohistochemical study of
three cases. Pathol Res Pract 1999; 195: 681-687.
Kuppers R, Brauninger A, Muschen M, et al.
Evidence that Hodgkin and Reed-Sternberg cells in Hodgkin disease do not
represent cell fusions. Blood 2001; 97: 818-821.
Lida S, Rao PH, Butler M, et al. Deregulation
of MUM1/IRF4 by chromosomal translocation in multiple myeloma. Nat Genet 1997;
17: 226-230.
Morente MM, Piris MA. REAL Classification. Una propuesta del
International Lymphoma Study Group para la clasificación de los linfomas. Patología 1995; 28: 319-358.
Muschen M, Rajewsky K, Brauninger A, et al. Rare occurrence of classical Hodgkin´s disease as a T cell lymphoma. J
Exp Med 2000; 191: 387-394.
Nakamura S, Nagahama M, Kagami Y, Yatabe Y, Takeuchi T, Kojima M, Motoori T,
Suzuki R, Taji H, Ogura M, Mizoguchi Y, Okamoto M, Suzuki H, Oyama A, Seto M,
Morishima Y, Koshikawa T, Takahashi T, Kurita S, Suchi T. Hodgkin's disease expressing follicular dendritic cell marker CD21
without any other B-cell marker: a clinicopathologic study of nine cases. Am J Surg Pathol 1999 Apr; 23(4): 363-76.
Oudejans JJ, Kummer JA, Jiwa M, et al. Granzyme
B expression in Reed-Sternberg cells of Hodgkin ´s disease. Am J Pathol 1996;
148: 233-240.
Pinkus GS, Pinkus JL, Langhoff E, et al. Fascin, a sensitive new marker for
Reed-Sternberg cells of Hodgkin ´s disease. Evidence for a dendritic or B cell
derivation? Am J Pathol 1997; 150: 543-562.
Quintanilla-Martinez L, Fend F, Moguel LR, Spilove L, Beaty MW,
Kingma DW, Raffeld M, Jaffe ES. Peripheral T-cell lymphoma with
Reed-Sternberg-like cells of B-cell phenotype and genotype associated with
Epstein-Barr virus infection. Am J Surg Pathol. 1999 Oct; 23(10): 1233-40.
Rudiger T, Ott G, Ott MM, et al. Differential diagnosis
between classic Hodgkin´s lymphoma, T-cell-rich B-cell lymphoma, and
paragranuloma by paraffin immunohistochemistry. Am J Surg Pathol 1998; 22:
1184-1191.
Schmid C, Pan
L, Diss T, Isaacson PG. Expression of B-cell antigens by Hodgkin's and Reed-Sternberg
cells. Am J Pathol. 1991 Oct; 139(4): 701-7.
Seitz V, Hummel M, Marafioti T,
Anagnostopoulos I, Assaf C, Stein H. Detection of clonal T-cell receptor
gamma-chain gene rearrangements in Reed-Sternberg cells of classic Hodgkin
disease. Blood. 2000 May 15; 95(10): 3020-4.
Seitz V, Hummel M, Anagnostopoulos I, Stein H. Analysis of BCL-6
mutations in classic Hodgkin disease of the B- and T-cell type. Blood. 2001 Apr
15; 97(8): 2401-5.
Siebert JD,
McClure SP, Banks PM, Gulley ML. Hodgkin's disease, mixed cellularity type,
with a B-cell immunophenotype. Report of a case and literature review. Arch
Pathol Lab Med. 1995 May; 119(5): 474-9.
Stoler MH, Nichols G, Symbula M, et al.
Lymphocyte predominance Hodgkin’s disease: evidence for a kappa light-chain
restriction monotypic B-cell neoplasm. Am J Pathol 1995; 146: 812-818.
von Wasielewski R, Mengel M, Fischer R, et al.
Classic Hodgkin´s disease: clinical impact of the immunophenotype. Am J Pathol
1997; 151: 1123-1130.
Vasef MA,
Alsabeh R, Medeiros LJ, Weiss LM. Immunophenotype of Reed-Sternberg and
Hodgkin's cells in sequential biopsy specimens of Hodgkin's disease: a
paraffin-section immunohistochemical study using the heat-induced epitope retrieval
method. Am J Clin Pathol.1997 Jul; 108(1): 54-9.
von Wasielewski R, Werner M, Fischer R, et al.
Lymphocyte predominant Hodgkin’s disease: an immunohistochemical analysis of
208 reviewed Hodgkin’s disease cases from the German Hodgkin Study Group. Am J
Pathol 1997; 150: 793-803.
Watanabe K, Yamashita Y, Nakayama A, et al. Varied B-cell immunophenotypes of Hodgkin/Reed-Sternberg cells in
classic Hodgkin´s disease. Histopathology 2000; 36;
353-361.
Ye
BH, Lista F, Lo Coco F, et al. Alterations of a zinc
finger-encoding gene, BCL-6, in diffuse large-cell lymphoma. Science 1993 ;
262: 747.
Zukerberg LR, Collins AB, Ferry JA, Harris NL.
Coexpression of CD15 and CD20 by Reed-Sternberg cells in Hodgkin's disease. Am
J Pathol. 1991 Sep; 139(3): 475-83.