
S5 Hallazgos inesperados en Inmunohistoquímica
Drs. Tomás Álvaro Naranjo /
Dr. Joaquín Jaén Martínez.
Servicio de Patología. Hospital Verge de la Cinta.
Tortosa.
C1. SÍNDROME
LINFOPROLIFERATIVO AUTOINMUNE
Historia clínica:
Niño magrebí de 8 meses de
edad que presenta febrícula, anemia, leucocitosis, hipergammaglobulinemia y adenopatías
múltiples.
Múltiples serologías
infecciosas negativas. Citometría de flujo de sangre periférica muestra
poblaciones linfocitarias normales. Mielograma no valorable (sin grumo). Se
realiza biopsia de ganglio linfático.
Estudio histológico:
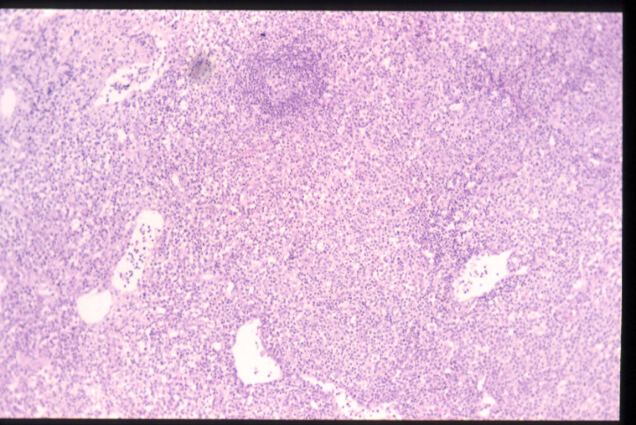
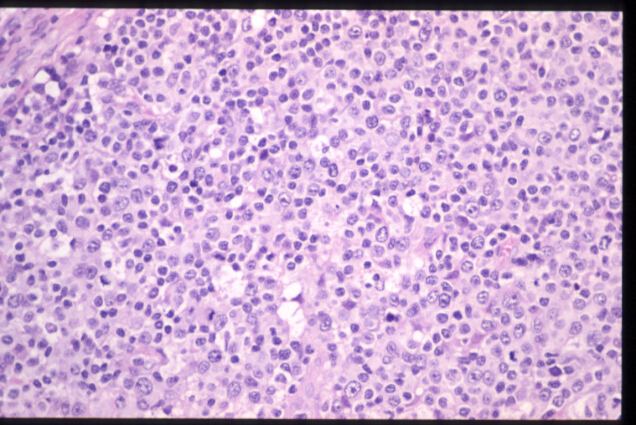
El estudio histológico mostró un
ganglio de mediano tamaño ocupado por una proliferación celular que se
distribuye por el espacio paracortical e interfolicular y que presenta una
morfología heterogénea, con elementos celulares de tamaño variable, predominio de
núcleos redondeados o con ligeras indentaciones, observación ocasional de
nucléolo en las células de mayor tamaño, abundante presencia de mitosis y una
cantidad amplia de citoplasma finamente granular. Se observan algunos centros
germinales, células plasmáticas y abundantes estructuras vasculares.
|
H.E
|
H.E. |
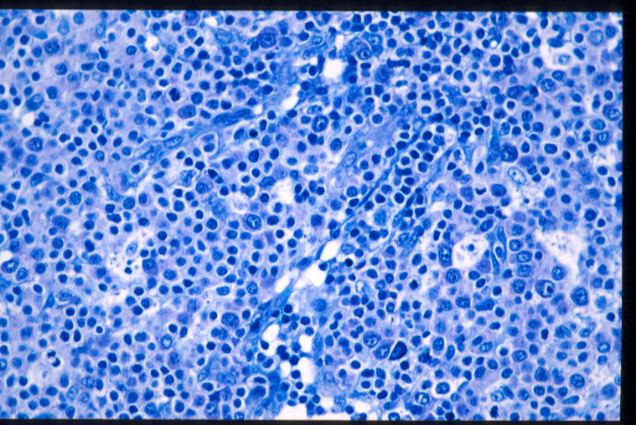
GIEMSA |
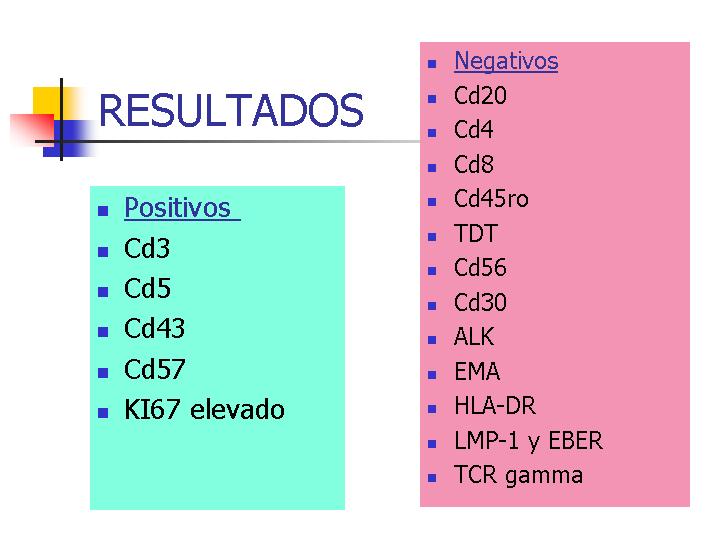
Estudio inmunohistoquímico:
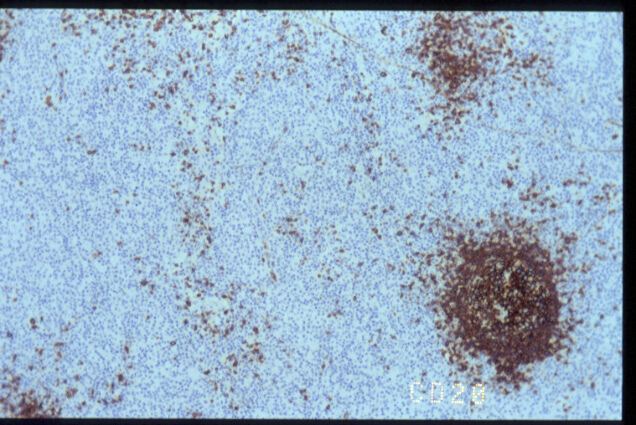
El estudio
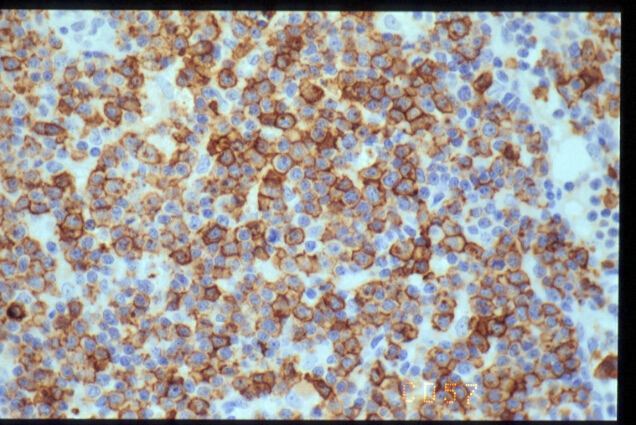
inmunohistoquímico mostró un inmunofenotipo positivo para CD3, CD5, CD43
y CD57. La población T resulta doblemente negativa para CD4 y CD8, y CD20 solo
pone de manifiesto restos de centros germinales atrapados. Otros marcadores
negativos son Tdt, CD56, CD30, ALK y EMA. El índice proliferativo evaluado con
Ki67 es alto. La investigación de la presencia de VEB fue negativa por métodos
IHQ (LMP-1) e HIS (EBER). El estudio con PCR para TCR-gamma fue negativo.
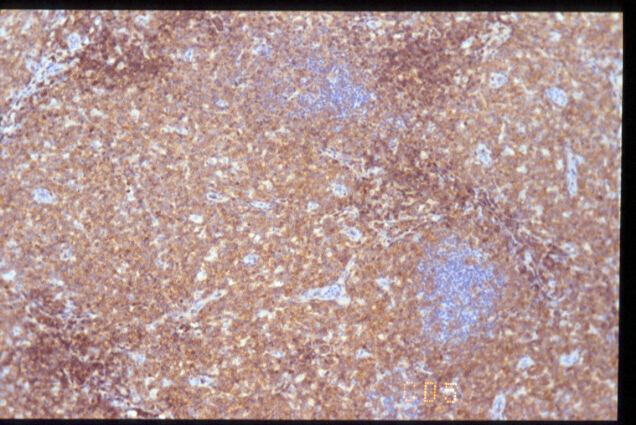
|
CD20
|
CD5
|
CD57
|
RESULTADOS
|
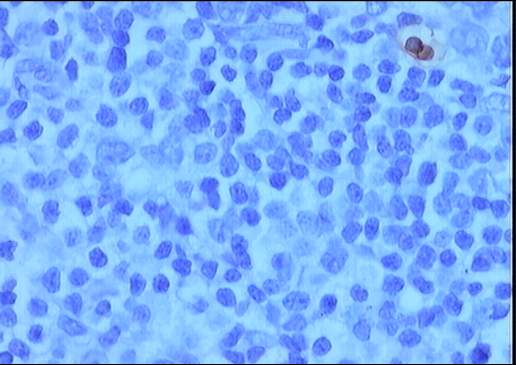
La
valoración del grado de apoptosis mediante TUNEL muestra un índice muy bajo,
tanto en la celularidad acumulada como en el interior de los centros germinales
residuales.
|
TUNEL |
Diagnóstico:
SÍNDROME
LINFOPROLIFERATIVO AUTOINMUNE
Comentarios al SLPA:
El
síndrome linfoproliferativo autoinmune (SLPA) resulta de un defecto en la
apoptosis de los linfocitos causado por mutaciones en el gen Fas, que codifica
el receptor para la apoptosis linfocitaria Fas/Apo-1/CD95 (Martin DA, et al. 1999). La enfermedad también puede deberse a mutaciones en
el gen que codifica el ligando de FAS (FAS-L, CD95L). Estas dos formas de la
enfermedad han sido referidas como SLPA tipo 1A y tipo 1B, respectivamente.
Adicionalmente existe un tipo II de SLPA causado por la mutación del gen de la
caspasa 10.
CD95
es una proteína transmembrana que pertenece a la familia del receptor TNF. La
interacción de CD95 con sus ligandos juega un papel fundamental en el control
de los linfocitos periféricos induciendo señales que conducen a la apoptosis.
Las mutaciones heterozigóticas de CD95 conllevan anomalías de la apoptosis
tanto de linfocitos normales como autoreactivos, que son los que se acumulan y
producen fenómenos autoinmunes en este síndrome.
La
enfermedad consiste en un cuadro crónico que se hereda de forma autosómica
dominante pero con un alto grado de variabilidad en su expresión clínica. El
síndrome se caracteriza por esplenomegalia, linfadenopatía,
hipergammaglobulinemia, autoinmunidad, linfocitosis B y la expansión de una
población inusual de células T, CD3+, CD4-CD8-, CD45RO-, CD45RA+, CD57+, que expresan el receptor alfa/beta de
células T.
|
|

Histopatológicamente
se caracteriza por preservación arquitectural, con hiperplasia folicular reactiva
y marcada expansión paracortical. Esta última puede ser tan intensa, junto a un
índice proliferativo tan alto, que sugieran fuertemente linfoma, lo que
justifica algunas interpretaciones erróneas (ver van der Werff T, et al, 1999)
y aconseja considerar la posibilidad de déficit del sistema Fas/Fas-L en niños
con síndromes linfoproliferativos atípicos inexplicados.
|
|

El
mecanismo fisiopatogénico que justifica este cuadro consiste en el acúmulo de linfocitos
T debido al fallo en la apoptosis mediada por FAS. Junto a esta parte
"linfoproliferativa" (término criticado por algunos autores), el
fallo en la apoptosis de los linfocitos B podría ser responsable de los
fenómenos autoinmunitarios que completan el síndrome.
|
|

La
definición de las bases moleculares de esta enfermedad no sólo permitió el
reconocimiento de la entidad (síndrome de Canale-Smyth), sino que mostró que las
mutaciones de FAS son compatibles con una supervivencia a largo término, ya que
estos pacientes llegan a la edad adulta, si bien con la posibilidad de
desarrollar un amplio abanico de enfermedades autoinmunes (entre las que
destacan la anemia hemolítica y trombocitopenia) y linfomas, además de
episodios intermitentes de linfadenopatía y esplenomegalia.
Recientemente
se ha señalado el hallazgo de déficit de apoptosis mediada por Fas en ausencia
de mutaciones del gen Fas como rasgo familiar predisponente al padecimiento de
enfermedades autoinmunes y cáncer (Ramenghi U, et al. 2000).
Agradecimientos
Agradecemos
la colaboración del Dr. Emilio Mayayo Artal, del servicio de Anatomía
Patológica del Hospital Joan XXIII, de cuyos archivos procede el material del
SLPA presentado.
Bibliografía
- Drappa,
J.; Vaishnaw, A. K.; Sullivan, K. E.; Chu, J.-L.; Elkon, K. B. : Fas gene
mutations in the Canale-Smith syndrome, an inherited lymphoproliferative
disorder associated with autoimmunity. New Eng. J. Med. 335:
1643-1649, 1996.
- Fisher,
G. H.; Rosenberg, F. J.; Straus, S. E.; Dale, J. K.; Middelton, L. A.; Lin,
A. Y.; Strober, W.; Lenardo, M. J.; Puck, J. M. : Dominant interfering Fas
gene mutations impair apoptosis in a human autoimmune lymphoproliferative
syndrome. Cell 81: 935-946, 1995.
- Illum N, Ralfkiaer E, Pallesen G, Geisler C Phenotypical and functional
characterization of double-negative (CD4-CD8-) alpha beta T-cell receptor
positive cells from an immunodeficient patient. Scand J Immunol
1991; 34: 635-45
- Infante AJ, Britton HA, DeNapoli T, et
al. The clinical spectrum in a large kindred
with autoimmune lymphoproliferative syndrome caused by a Fas mutation that
impairs lymphocyte apoptosis. J Pediatr 1998; 133: 629-633.
- Krammer,
P. H. : CD95's deadly mission in the immune system. Nature 407:
789-795, 2000.
- Lim MS, Straus SE, Dale JK, Fleisher TA, Stetler-Stevenson M,
Strober W, Sneller MC, Puck JM, Lenardo MJ, Elenitoba-Johnson KS, Lin AY,
Raffeld M, Jaffe ES. Pathological findings in human autoimmune
lymphoproliferative syndrome. Am
J Pathol 1998;153:1541-50
- Martin
DA, Zheng L, Siegel RM, et al. Defective
CD95/APO-1/Fas signal complex formation in the human autoimmune
lymphoproliferative syndrome, type Ia.
Proc Natl Acad Sci USA 1999;
96: 4552-4557.
- Nagata,
S. : Human autoimmune lymphoproliferative syndrome, a defect in the
apoptosis-inducing Fas receptor: a lesson from the mouse model. J.
Hum. Genet. 43: 2-8, 1998.
- Ramenghi U, Bonissoni S, Migliaretti G, et al. Deficiency of the
Fas apoptosis pathway witout Fas gene mutations is a familial trait
predisposing to development of autoimmune diseases and cancer. Blood 2000;
95: 3176-3182.
- Rieux-Laucat,
F.; Le Deist, F.; Hivroz, C.; Roberts, I. A. G.; Debatin, K. M.; Fischer,
A.; de Villartay, J. P. : Mutations in Fas associated with human
lymphoproliferative syndrome and autoimmunity. Science 268:
1347-1349, 1995.
- Sneller MC, Wang J, Dale JK et al. Clinical, inmunologic, and
genetic features of an autoimmune lymphoproliferative syndrome associated
with abnormal lymphocyte apoptosis. Blood 1997; 89: 1341-1348.
- Vaishnaw,
A. K.; Orlinick, J. R.; Chu, J.-L.; Krammer, P. H.; Chao, M. V.; Elkton,
K. B. : The molecular basis for apoptotic defects in patients with CD95
(Fas/Apo-1) mutations. J. Clin. Invest. 103: 355-363, 1999.
13. van der Werff T, Bosch J, Delabie J, et al. Revision of the diagnosis of T-zone lymphoma in the father of a patient with autoimmune lymphoproliferativesyndrome type II. Br J Haematol 1999; 106: 1045-1048.