VIRUS Y NEOPLASIAS HEMATOLÓGICAS
Dra. Estel·la Matutes, MD, PhD, MRCPath
Senior Lecturer and Consultant Haematologist
Academic Department of Haematology and Cytogenetics
The Royal Marsden Hospital. London
INTRODUCCIÓN
RETROVIRUS
Mecanismos oncogénicos de los retrovirusEpidemiología
HTLV-I
HTLV-II
HIVVirus de la Leucemia Linfoma T (HTLV-I): LLTA
Virus de la leucemia humana tipo II (HTLV-II)
Virus de la inmunodeficiencia adquirida (VIH)Etiopatogenia y Epidemiología
Estructura genómica y detección del VEB
Patología asociada al VEB- Linfoma de Burkitt
- Linfomas B en individuos con inmunodeficiencia
- Linfoma angiocéntrico nasal tipo T/NK
- Leucemia de linfocitos granulares (LLG)- NK
- Enfermedad de Hodgkin (EH)
- Otros procesosHHV8/KSHV o virus herpes asociado al sarcoma de Kaposi
Etiopatogenia y epidemiología
Patología asociada al HHV8- Sarcoma de Kaposi
- Linfoma primario de cavidades (BCBL o PEL)
- Enfermedad de Castleman
- Otros procesos asociados a HHV8OTROS VIRUS
Virus de la hepatitis C (VHC)
HHV6
CONCLUSIONES
BIBLIOGRAFÍA
El papel directo o indirecto de ciertos virus en el desarrollo de neoplasias hematológicas ha sido intuido desde principios de este siglo en base a modelos animales. Si bien, no ha sido hasta las dos últimas décadas durante las cuales se ha demostrado de una forma concluyente la implicación causal de una variedad de virus en el desarrollo de determinados tipos de leucemias y linfomas en el ser humano.
Los virus con potencial oncogénico en el hombre pueden, en líneas generales, clasificarse en dos grandes categorías: 1- los retrovirus (virus RNA) y 2- los virus del grupo herpes (DNA) y dentro de cada grupo se identifican diferentes subtipos (Tabla 1).
Tabla 1- Virus con potencial oncogénico:
Retrovirus:
Virus del grupo herpes:
Otros posibles:
|
La identificación del primer retrovirus capaz de producir una leucemia en animales fue documentada en 1910 por el Dr. Roux que demostró el papel de un retrovirus en el desarrollo de la leucemia en pollos y, por ello, fue designado retrovirus de la leucemia aviar. Si bien, no fue hasta la década de los 50 cuando el campo de la retrovirología adquirió de nuevo interés culminando con el hallazgo de un número de retrovirus como agentes causales de leucemias en animales así como de otros procesos no tumorales, como aplasia e inmunodeficiencia. A finales de la década de los 70 y principios de los 80 se descubre el primer retrovirus causante de un tipo particular de leucemia linfoma T en el hombre y por ello se le designó virus de la leucemia/linfoma T humana (HTLV-I). Más tardíamente, se describieron otros retrovirus, esencialmente el HTLV-II que posee una homología en secuencia nucleótida de un 60% al compararse con el HTLV-I y, finalmente, el virus de la inmunodeficiencia adquirida (VIH).
Mecanismos oncogénicos de los retrovirus
En base al potencial oncogénico y al mecanismo de desarrollo de la neoplasia, los retrovirus se clasifican en dos grandes grupos:
1- retrovirus que contienen un oncogén y causan una leucemia aguda ("acute leukaemi virus")
2- retrovirus que no poseen un oncogén y causan leucemias crónicas ("chronic leukaemia virus").
Los primeros, al integrarse en la célula huésped, expresan el oncogén, dando lugar al desarrollo de un cuadro agudo. La integración del retrovirus en el tumor no es clonal ya que la expresión del oncogén es independiente del lugar del genoma de la célula huésped en donde el retrovirus se inserte. Por el contrario, los retrovirus que no poseen oncogén, se integran en la célula huésped en un punto clave y único, adyacente a un gen que interviene en el proceso de multiplicación o diferenciación celular al cual disregulan dando lugar al origen de una célula neoplásica. Por tanto, la integración de estos virus es clonal en el tumor.
El HTLV-I es un retrovirus especial en el sentido de que no posee oncogén como los agudos ni tampoco se integra en un sitio clave del genoma como ocurre con los crónicos. Si bien la integración del HTLV-I es clonal en un tumor en particular, varía el punto donde se inserta de un tumor a otro. Aunque se desconoce el mecanismo por el cual el HTLV-I actúa, se cree que éste es a distancia ("trans-acting") mediante su secuencia px o tax que da lugar a la disregulación y activación de una serie de genes celulares tales como los que codifican para diferentes interleukinas (IL) básicamente: IL-2, IL-3 y IL-6, factores de necrosis tumoral (TNF), oncogenes celulares (c-myc, c-fos) etc. (1, 2). Asimismo se ha especulado que el tax en las primeras fases de leucemogénesis interfiera con mecanismos de reparación de ADN celular dando lugar a una inestabilidad cromosómica. Un punto importante a considerar es el de que, si bien el HTLV-I es el primer factor inicial desencadenante de la leucemia linfoma T del adulto (LLTA), no es suficiente "per se" para su desarrollo, ya que sólo un 0.5-1% de portadores del virus desarrollan leucemia. Es necesario el concurso de factores secundarios que, en la actualidad, son desconocidos. El sistema inmunitario del individuo parece jugar, asimismo, un papel importante ya que se ha demostrado que individuos infectados por el HTLV-I poseen clonas citotóxicas anti-HTLV-I capaces de eliminar células infectadas por dicho retrovirus.
Al contrario que el HTLV-I, se desconocen enfermedades oncológicas asociadas al HTLV-II habiéndose demostrado solamente que este retrovirus podría estar vinculado a unos pocos casos de síndromes linfoproliferativos.
En cuanto al papel del VIH en el desarrollo de neoplasias parece ser indirecto y multifactorial originado por el estado profundo de inmunosupresión del individuo infectado que permite el desarrollo de neoplasias, en su gran mayoría asociadas a otros virus, en especial del tipo herpes.
Estudios serológicos iniciales demostraron que el HTLV-I es endémico en Japón, especialmente en la región Sudoeste y en poblaciones de ancestro africano de las islas del Caribe. Durante la década de los 80 y principios de los 90 se ha demostrado que este retrovirus se halla mucho más extendido, documentándose una seroprevalencia alta así como su patología asociada en: África Central y Oeste, Irán -en particular en una población judía emigrante de Israel-, Sudamérica -especialmente en Brasil, Perú, Chile, Colombia y en menor proporción en Argentina y Venezuela-, Melanesia y, en poblaciones emigrantes de estos lugares a Europa y Estados Unidos (1, 3). No existe un foco evidente en Europa aunque es probable que el virus se halle en algunos países tales como Rumania. En Espańa, el HTLV-I y la LLTA se han observado, esencialmente, en unos pocos pacientes de origen latinoamericano.
Los mecanismos de transmisión del HTLV-I básicamente son: la vía horizontal de madre a nińo a través de la leche materna, transfusión de productos celulares o inoculación con jeringuilla y, con menor frecuencia, la vía sexual de varón a mujer. La instauración de medidas preventivas en algunos países tales como el screenning sistemático de donantes de sangre y/o evitar el amamantamiento de bebés en madres seropositivas ha sido el determinante de un descenso de la seroprevalencia por HTLV-I en algunos países endémicos como Japón.
El HTLV-II se aisló en 1982 a partir de una línea celular de un bazo procedente de un paciente que había sido diagnosticado de tricoleucemia en una forma variante T. Debido a que tal proceso no se halla establecido ni reconocido como entidad, es muy probable que el paciente se hallara afecto de otro proceso linfoproliferativo T.
El HTLV-II posee una distribución mundial muy diferente al del HTLV-I. El HTLV-II es endémico en tribus amerindias de América del Sur y Central en donde la seroprevalencia puede alcanzar niveles de hasta un 30% así como en pigmeos africanos del Zaire y Camerún. En países desarrollados el HTLV-II se detecta casi exclusivamente en drogadictos, en su mayoría coinfectados con VIH. Los mecanismos de transmisión del HTLV-II son similares a los del HTLV-I si bien, debido a que su distribución es muy diferente a la del HTLV-I, el uso de jeringuillas y transfusión son las principales vías para la transmisión del HTLV-II.
Es bien conocida la distribución mundial del VIH que afecta, principalmente aunque no de forma exclusiva, a individuos homosexuales, drogadictos y hemofílicos entre otros grupos sectarios. Las formas de transmisión del VIH son múltiples pero las más frecuentes y efectivas son la sexual y endovenosa a través de jeringuilla o transfusión.
El VIH se halla vinculado al desarrollo de una
gran variedad de procesos oncológicos de origen o no hematológico. Las causas o
"background" por las que el VIH predispone al desarrollo de neoplasias linfoides
y no linfoides probablemente sean multifactoriales, ya que el VIH no se halla integrado en
las células neoplásicas. Otros virus, en particular del grupo herpes: VEB y HHV8, juegan
un papel fundamental en el desarrollo del tumor. Así, la gran mayoría de linfomas
primarios del sistema nervioso central poseen integrado el EBV y, hasta un 40%, el HHV8.
Entre los mecanismos involucrados en el desarrollo del tumor derivado de la infección por
VIH y, probablemente mediados a través de su gen tat, destacan: 1) la disregulación en
la producción de citoquinas, tales como IFN-alfa e hipersecreción de IL-6 que conlleva
la estimulación de linfocitos B y de IL-10 y
2) promover el crecimiento y proliferación de células endoteliales, factor que
probablemente juega un papel en el desarrollo del sarcoma de Kaposi, neoplasia altamente
asociada al VIH. Otras proteínas y/o oncogenes tales como el c-myc, bcl-6 y mutaciones de
p53 se hallan involucrados en los linfomas en individuos VIH+ de forma similar a lo que
acontece en individuos que son HIV negativos.
Tabla 2- Patología asociada a retrovirus
| HTLV-I:
Leucemia linfoma T del adulto
(LLTA) Procesos inflamatorios de naturaleza inmune Otros (ej. inmunodeficiencia) HTLV-II: No definida VIH (asociados o no a VEB y HHV8): |
Virus de la Leucemia Linfoma T (HTLV-I): LLTA
El HTLV-I es el agente causal de un tipo característico de linfoma/leucemia designado leucemia linfoma T del adulto (LLTA). Asimismo, el HTLV-I se halla involucrado en el desarrollo de una variedad de patologías inflamatorias tales como la paraparesia espástica tropical, uveítis, artritis, polimiositis, dermatitis etc., las cuales probablemente tienen una base inmune, ya que el virus no se halla integrado de una manera clonal (1).
La LLTA es un síndrome linfoproliferativo maduro T que podría catalogarse dentro de los síndromes leucemia/linfoma o linfomas T con expresión leucémica. Con gran frecuencia los pacientes manifiestan un cuadro leucémico, si bien el origen del tumor sea probablemente nodal. De acuerdo a las manifestaciones clínicas se distinguen: la forma leucémica (75% de los casos) que incluye tres modalidades: aguda (65%), crónicas y latentes (10%) y la forma linfoma sin expresión leucémica (25%) (4). La hipercalcemia es un rasgo clínico característico y es el resultado de la producción de una proteína similar a la hormona paratiroidea secretada por las células infectadas por el HTLV-I. El curso clínico es agresivo con refractariedad al tratamiento (4).
El cuadro morfológico de sangre periférica es típico en la LLTA. Suele observarse pleomorfismo en cuanto a tamańo celular y contornos nucleares, siendo el linfocito "en flor" con un núcleo marcadamente irregular la forma celular más típica (Fig. 1). Por el contrario, no existe un patrón histológico único asociado a la LLTA, por lo que en la clasificación REAL la LLTA se define por la presencia del retrovirus. En el ganglio la infiltración es difusa y el pleomorfismo, al igual que en sangre periférica, es marcado (Fig. 2). El patrón difuso mixto con presencia de células de varios tamańos es el más habitual, pero también puede manifestarse como un linfoma de células grandes, inmunoblástico o incluso de células pequeńas. Han sido documentados, infrecuentemente, casos con un patrón "Hodgkin's like" y/o de linfoma anaplásico. El patrón histológico de la piel no es específico en la LLTA igual que ocurre en el ganglio linfático. En general, se observa una infiltración dérmica por linfocitos de núcleo irregular, pero puede hallarse presente epidermotropismo con un patrón similar al de la micosis fungoides lo cual plantea problemas de diagnóstico diferencial con linfomas cutáneos T.
El inmunofenotipo es el de un linfocito T maduro (TdT-, CD2+, CD5+, CD7-) con baja expresión del CD3 y, en la mayoría de los casos, CD4+, CD25+. Este último marcador, que detecta el receptor alfa de la IL-2, resulta de la infección por HTLV-I tanto en portadores como en enfermos con leucemia.
No existe una alteración citogenética única en la LLTA si bien la presencia de cariotipos complejos es frecuente en las formas aguda y linfoma.
El HTLV-I se halla integrado de forma clonal en todos los casos de LLTA, lo cual corrobora su papel etiológico primario en el desarrollo de la neoplasia; el receptor de células T (RCT) se halla reordenado.
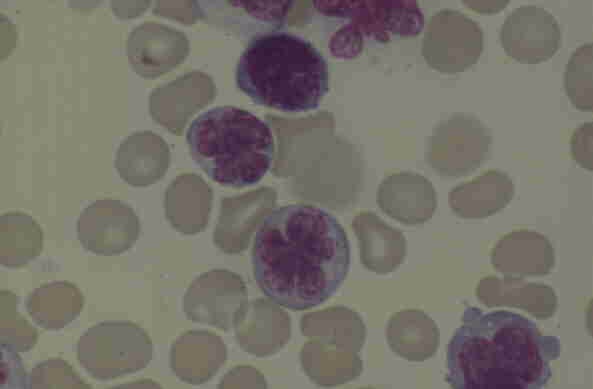 |
Figura 1- Linfocitos circulantes en un caso de leucemia linfoma T del adulto (LLTA) mostrando un núcleo irregular, típico de célula en flor. |
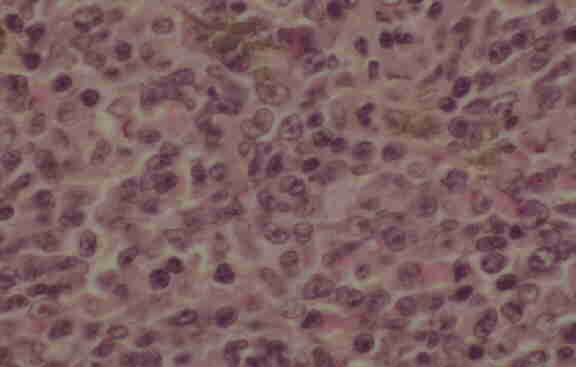 |
| Figura 2- Corte de un ganglio en una LLTA-modalidad linfoma-. El patrón de infiltración es difuso con predominio de células grandes, cromatina reticular y nucléolos prominentes. |
Virus de la leucemia humana tipo II (HTLV-II)
No se ha establecido de forma concluyente un vínculo entre el HTLV-II y un tipo de neoplasia humana. Tras su identificación en la línea celular, ha sido documentada mediante ELISA una serología positiva para el HTLV-II en una proporción de pacientes con leucemia de linfocitos granulares (LLG) así como, mediante PCR, se han detectado secuencias de HTLV-II en las células de unos pocos individuos con esta leucemia. Si bien, estudios posteriores aplicando Western-blott no han confirmado la especificidad de los resultados serológicos por ELISA hallándose patrones indeterminados (5). Asimismo, tampoco se ha podido demostrar la integración clonal del HTLV-II en los linfocitos circulantes de los pacientes con LLG. Por consiguiente, es muy probable que el HTLV-II no juegue un papel etiológico importante en el desarrollo de la LLG y que tal vez estos hallazgos sean el reflejo de una reacción y/o activación viral, aunque la posibilidad de que otros retrovirus relacionados con el HTLV-II se hallen involucrados en el desarrollo de esta leucemia no queda descartada.
Virus de la inmunodeficiencia adquirida (VIH)
Los procesos neoplásicos asociados al VIH son múltiples y nos referiremos sólo a los del sistema linfoide o a aquellos en que otros virus se hallan involucrados. Entre éstos destacan: linfomas no Hodgkin sistémicos y primarios del sistema nervioso central, enfermedad de Hodgkin (EH), linfomas primarios de cavidades (BCBL o PEL), enfermedad de Castleman y sarcoma de Kaposi. Aunque este último no puede considerarse un proceso hematológico, debido a su estrecha asociación al VIH y al HHV8 también se discutirá.
La mayoría de linfomas en los individuos VIH+ presentan coinfección con el VEB y son de origen B. El VEB se detecta prácticamente en todos los casos de linfomas primarios de sistema nervioso central (6) y, con menor frecuencia, en los linfomas sistémicos. La demostración de la integración clonal del VEB en estos linfomas sustenta su papel etiológico en el desarrollo de los mismos. Clínicamente son agresivos y de difícil manejo, con una corta supervivencia. Desde un punto de vista histológico, la mayoría corresponden a linfomas de células grandes, generalmente de tipo inmunoblástico y/o centroblástico y, con menor frecuencia, anaplásico o bien se encuadran dentro del tipo linfoblástico-Burkitt (Figuras 3, 4, 5). No es infrecuente que existan dificultades en su clasificación dentro de la REAL ya que no es rara la presencia de una marcada diferenciación plasmocitoide (7) y/o manifestar características de los diferentes subtipos de linfomas en un mismo tejido (8).
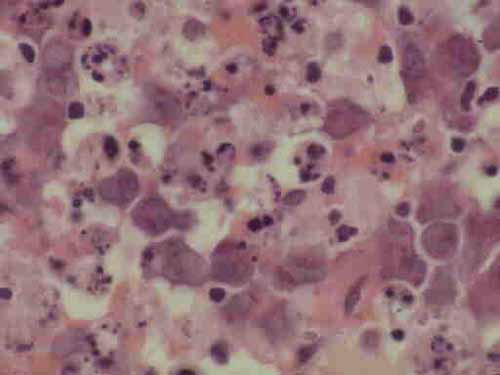 |
| Figura 3- Corte de un ganglio en un paciente con VIH y linfoma centroblástico donde se aprecia una marcada apoptosis. |
 |
| Figura 4- Inmunohistoquímica del ganglio de la Figura 3 mostrando que la mayoría de las células son CD20+. |
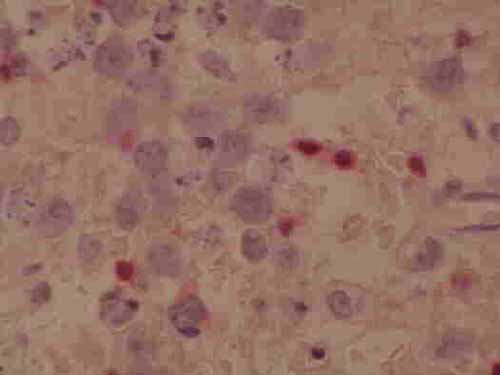 |
| Figura 5- Inmunohistoquímica del ganglio de la Figura 3 mostrando reactividad para el CD3 en linfocitos pequeńos mientras que las células grandes son negativas. |
El inmunofenotipo es B, en general SmIg+, CD19+, CD20+, CD22+ (9), si bien los anaplásicos Ki-1+ pueden poseer un fenotipo indeterminado. El gen que codifica para las cadenas pesadas de la Ig se halla reordenado en la mayoría de los linfomas (10); en algunos casos, éste puede hallarse en línea germinal si bien se demuestra clonalidad para el VEB. La presencia del VEB es más frecuente en el subtipo inmunoblástico que en los restantes tipos histológicos.
Los individuos con VIH poseen también una mayor incidencia de enfermedad de Hodgkin (EH). Clínicamente, ésta suele comportarse de modo más agresivo que en los individuos VIH negativos, presentándose en estadios avanzados (III y IV) y con frecuente afectación extranodal, tal como médula ósea. Los subtipos más frecuentes son la celularidad mixta y deplección linfocítica.
Los linfomas primarios de cavidades, la enfermedad de Castleman y el sarcoma de Kaposi se describen dentro de la patología asociada al HHV-8
El segundo grupo de virus con potencial oncogénico lo constituye el del grupo herpes. Entre éstos, el prototipo es el virus de Epstein Barr (VEB). El VEB se halla mundialmente distribuido, al contrario que el HTLV-I/II que posee focos de endemicidad. La infección primaria acontece en la adolescencia y es asintomática o produce un cuadro viral, la mononucleosis infecciosa. El individuo infectado mantiene anticuerpos anti-VEB de tipo IgG y el virus queda acantonado sustancialmente en la nasofaringe, en las células epiteliales y en los linfocitos B. El receptor para el VEB es la molécula CD21.
La asociación del VEB con el carcinoma de nasofaringe se sospechó desde hace más de dos décadas pero sólo, "a posteriori", pudo demostrase su papel etiopatogénico, no sólo en esta neoplasia, sino también en el linfoma de Burkitt endémico.
Estudios epidemiológicos, serológicos y moleculares han demostrado que el VEB posee un papel importante en el desarrollo de una variedad de procesos linfoides de naturaleza B y T así como de la EH y linfomas que se desarrollan en individuos inmunodeprimidos, tales como portadores de VIH o post-trasplante (Tabla 3).
Estructura genómica y detección del VEB
El genoma (ADN) del VEB codifica para un número de proteínas, las cuales poseen una función especifica. Entre las más importantes destacan tres proteínas latentes de membrana (LMP), dos que codifican ARN nucleares (EBER1/2) y seis que codifican factores de transcripción (EBNAs). La EBNA1 es esencial para el mantenimiento del virus, mientras que la LMP-1 posee capacidad oncogénica y se desconoce la función de las EBERs.
La expresión de las diferentes proteínas en células normales y neoplásicas es diferente. Se han descrito tres tipos de expresión o latencia que, hasta cierto punto, permiten eludir el control inmunitario del individuo mediado por los linfocitos citotóxicos. En todos los individuos, con o sin tumores, el EBNA-1 se halla expresado. El tipo de latencia I es típico del linfoma Burkitt y se caracteriza por la expresión únicamente de EBER1/2. El tipo II, característico del carcinoma de nasofaringe, EH y, hasta cierto punto, de linfomas angiocéntricos, particularmente nasales, se caracteriza por la expresión de LMP1/2 además de EBER1/2. El tipo de latencia III, que se observa en procesos linfoides en individuos con inmunodeficiencia y en las líneas celulares linfoblastoides, muestra un patrón tal en el que todas las proteínas se hallan expresadas.
La detección del VEB puede realizarse mediante hibridación in situ (HIS), aplicada esencialmente para la detección de EBERs, inmunohistoquímica con anticuerpos anti-LMP-1/2 y EBNA-1, Southern blot que permite demostrar clonalidad y PCR.
Patología asociada al VEB
Tabla 3 - Patología asociada al virus de Epstein Barr (VEB)
Linfoide de origen B:
Linfoide de origen T/NK
Otros:
VEB como agente secundario y/o debatida su participación:
|
El VEB se asocia al linfoma de Burkitt en más del 95% de pacientes procedentes de zonas endémicas, ej. África, mientras que el virus se detecta sólo en una minoría de casos de linfoma de Burkitt no endémico. La infección por malaria se considera un cofactor importante que incrementa la susceptibilidad del individuo infectado por el virus para el desarrollo del tumor.
En el linfoma de Burkitt es característica la translocación t(8;14)(q24;q32) o una de sus variantes t(2;8) o t(8;22) que dan lugar al reordenamiento del oncogén c-myc (8q24) con el gen que codifica la cadena pesada o ligera de la Ig, determinando una disregulacion de la misma. El hecho de que el VEB no se detecte en linfomas Burkitt en países no endémicos y que, por otro lado, éstos muestren la misma alteración molecular, ej. t(8;14), indica que otros factores no relacionados con el VEB dan lugar a la misma alteración y desarrollo de un tumor idéntico. Por consiguiente, el VEB no es imprescindible para el desarrollo de un linfoma de Burkitt.
Si bien ambos tipos de linfoma de Burkitt, endémico VEB+ y no endémico VEB -, poseen una base genética idéntica y patrones citológicos (LLA-tipo L3 o linfoma linfoblástico con un patrón en cielo estrellado) e inmunofenotípicos (célula madura B con expresión de inmunoglobulinas y TdT-) similares, las características clínicas son hasta cierto punto diferentes. El Burkitt endémico afecta a individuos más jóvenes y suele presentarse como masas extranodales, mientras que la afectación ganglionar y de médula ósea es más común en el Burkitt no endémico. En este último, y principalmente cuando incide en adultos, debe siempre sospecharse y descartarse que el linfoma de Burkitt no sea el resultado de la transformación de un linfoma folicular.
Éstos se observan, esencialmente, en pacientes que han sido sometidos a transplante de órganos y reciben drogas inmunosupresoras así como en individuos infectados por el VIH (estos últimos se han discutido dentro del apartado de patología asociada al VIH). En cuanto a los linfomas post-trasplante, la proliferación linfoide inicialmente es policlonal y, en algunos enfermos, si se suspende la terapia inmunosupresora el linfoma puede regresar. En su mayoría son linfomas de células grandes (Fig. 6) y el tipo de latencia del VEB es la II.
El VEB se halla íntimamente implicado en la etiopatogenia de los linfomas angiocéntricos, particularmente en los nasales de origen T o de células natural killer (NK).
Los linfomas nasales angiocéntricos NK/T se observan, con preferencia, en pacientes de origen oriental, ej. procedentes de China, Japón (11, 12). También se ha documentado una prevalencia elevada en ciertos países latinoamericanos tales como Perú, Guatemala, etc. (13), lugares en donde algunos de los pacientes tienen un ancestro Maya. Son más raros los casos documentados en Europa y Estados Unidos. Estos linfomas predominantemente afectan las cavidades nasales y orofaringe, aunque pueden manifestarse o extenderse a otros órganos tales como piel, tracto digestivo, bazo, glándulas salivales, etc. Su curso clínico es agresivo.
La histología muestra un infiltrado pleomórfico linfocitario con marcada angiocentricidad (Fig. 7). Las células son de tamańo mediano o grande, poseen un núcleo irregular y, con frecuencia, nucléolos prominentes. Se ha descrito una forma blástica o blastoide de linfoma nasal que es poco frecuente y que, a diferencia de la forma típica, no se asocia al VEB (11, 12).
En base a las características inmunofenotípicas y genotípicas, se distinguen dos subtipos: 1) linfoma angiocéntrico NK, en el que las células poseen un fenotipo CD2+, CD56+ (Fig. 8) y son negativas para la mayoría de antígenos de diferenciación T, incluyendo el CD3, si bien expresan la cadena épsilon del CD3, y 2) linfoma T, en el que las células además de expresar el CD2 y CD56 son positivas para una variedad de marcadores T. En ambos subtipos, las células son TIA-1+, marcador que identifica proteínas asociadas a los gránulos de los linfocitos citotóxicos y células NK y también perforinas (14). El receptor de células T (RCT) se halla en línea germinal en el subtipo NK mientras que el subtipo T reordena la cadena g del RCT.
El VEB se halla presente, en la mayoría de los casos, en una forma de expresión de latencia tipo II similar a la de la EH y carcinoma nasofaríngeo (EBNA1+, LMP1/2+, EBNA2-, EBERs+) (Fig. 9). En algunos casos se ha documentado que dentro de un mismo tumor existe cierta heterogeneidad con tipos de latencia II (LMP-1+) y I (LMP1-) (15). La demostración mediante Southern-blot con sondas específicas para el VEB de una sola banda, ocasionalmente dos bandas, en estos tumores no sólo ha permitido demostrar su clonalidad sino también el papel fundamental que posee el VEB en esta neoplasia.
El VEB ha sido documentado en una pequeńa proporción de casos de LLG-NK. Éstos se presentan en su forma de linfoma o leucemia, poseen un curso agresivo y son más frecuentes en países orientales, principalmente Japón, aunque también se han descrito en países occidentales (16). El cuadro clínico, a veces, puede remedar histiocitosis maligna inducida por un proceso viral.
Estudios recientes incluyendo un número elevado de casos, sin embargo, demuestran que el VEB se halla involucrado en una minoría de los mismos y, por consiguiente, no es un factor esencial para el desarrollo de la LLG tipo NK (17).
La presencia del VEB en la LLG-NK se ha demostrado mediante ISH e immunoblotting. Por el contrario, la forma más común de LLG, de origen T, no se halla asociada al VEB.
El VEB se detecta en aproximadamente el 40% de casos de EH en países occidentales. Su presencia es mucho más frecuente en adolescentes y nińos donde puede alcanzar una incidencia del 80% y, con preferencia, se observa en el subtipo histológico celularidad mixta aunque también se ha descrito en los otros subtipos, pero muy raramente en el predominio linfocítico. Más raramente el VEB se ha detectado en casos excepcionales de EH que se desarrollan post-trasplante de órgano. Estudios de casos familiares de EH no han demostrado que exista un nivel de concordancia en cuanto a serología para el VEB y expresión de EBERs en el tejido de dichos individuos. Por consiguiente, el VEB parece no poseer un papel importante en el desarrollo de EH familiar (18).
El tipo de latencia del VEB en la EH es el II. La presencia de VEB en la EH se ha demostrado por ISH, Southern-blot e inmunohistoquímica empleando anticuerpos monoclonales (AcMo) anti-LMP2A. Esta proteína se identifica en las células de Hodgkin y de Reed-Sternberg con un patrón esencialmente granular asociado a la membrana o en el área de Golgi (19).
El mecanismo patogénico del VEB en la EH se desconoce. La demostración de su integración clonal en las células de Reed-Sternberg sustenta no sólo el origen clonal de las mismas sino también que la EH se ha desarrollado en una célula infectada por el VEB.
Se ha sugerido que el VEB podría ser el agente causal del linfoma T angioinmunoblástico. Estudios iniciales demostraron, por Southern blot, la presencia del VEB en un paciente y, posteriormente, se ha documentado la presencia de VEB mediante PCR en el tejido de la gran mayoría de pacientes con linfoma angioinmunoblástico (20). Sin embargo, el análisis simultáneo mediante ISH e inmunohistoquímica ha permitido objetivar que el VEB se halla integrado en las células B o en ambos tipos de linfocitos B y T (21). Por consiguiente, es muy probable que el VEB no sea un factor primario etiológico del linfoma angioinmunoblástico sino más bien una consecuencia del proceso neoplásico en un individuo con un sistema inmunitario alterado que permita la expansión policlonal de células infectadas por el VEB.
Asimismo, el papel etiológico del VEB en el linfoma Ki-1 se halla debatido. Los casos documentados VEB+ mediante ISH e inmunohistoquímica no poseen un fenotipo T sino B o nulo, lo cual no se corresponde con la definición de linfoma Ki-1 que en su mayoría son de origen T. Es, por consiguiente, posible que entre estos casos se incluyan linfomas de célula grande.
HHV8/KSHV o virus herpes asociado al sarcoma de Kaposi
El HHV8 es un virus del grupo herpes tipo gamma-2 que se identificó inicialmente en 1994 en el sarcoma de Kaposi en individuos infectados por el VIH y con síndrome de inmunodeficiencia adquirida (SIDA) y por ello se le designó KSHV o virus del sarcoma de Kaposi (22).
En base a su estructura molecular, el HHV8 pertenece a la familia de los virus gammaherpes virinae y posee una secuencia nucleótida con gran homología a la del VEB. El HHV8 se caracteriza por su marcado linfotropismo. En los linfocitos, el HHV8 se halla integrado en su forma latente pero, bajo estímulos adecuados, el virus pasa a su forma activa y productiva con formación de partículas virales cuya liberación conlleva la infección de otros elementos celulares así como la regulación de una variedad de genes celulares. Aunque este virus se detectó inicialmente mediante técnicas moleculares, "a posteriori" ha sido posible su cultivo y visualización mediante estudios ultraestructurales. Parece ser que existe una cierta interacción entre el HHV8 y el VEB inhibiendo este último la replicación del primero. Asimismo, se ha especulado que la frecuencia elevada de sarcomas de Kaposi en individuos con VIH podría deberse a que el VIH ejerce un efecto promotor a través de la proteína tat para el desarrollo de las lesiones cutáneas y vasculares al estimular la proliferación vascular, si bien el VIH no se considera un cofactor esencial.
Estudios de secuenciación del HHV8 han demostrado que su ADN codifica para una variedad de proteínas afines a proteínas celulares y de importancia en el ciclo, en la diferenciación y activación celular y en la apoptosis. Entre otras destacan: a) un homólogo de la IL-6, que posee características funcionales similares a la IL-6 endógena y que, por tanto, podría jugar un papel en la patogenia de los procesos asociados a HHV8 a través de la estimulación de células linfoides (23), b) una ciclina con similitudes a la ciclina-D2 humana y c) un homólogo del oncogén bcl-2.
Se desconoce la distribución mundial del HHV8 en la población sana, aunque se especula que se halla probablemente presente en una gran mayoría de individuos normales a niveles no detectables. Estudios en un limitado número de casos muestran una gran variación, con incidencias de individuos seropositivos de menos del 5% en Inglaterra, 25% en Estados Unidos y 50% en Uganda.
La detección de anticuerpos anti-HHV-8 puede realizarse mediante ELISA, inmunofluorescencia y Western-blot. Estudios serológicos, utilizando un test de inmunofluorescencia indirecta, demostraron la presencia de anticuerpos anti-HHV8 en la mayoría de pacientes infectados con el VIH y sarcoma de Kaposi, mientras que un grupo relativamente reducido de donantes de sangre en USA fue negativo. El grado de especificidad del test, sin embargo, es incierto, ya que se han documentado patrones de positividad intermedio en individuos control de origen africano (Uganda) y en Italia. La seroconversión con presencia de anticuerpos anti-HHV8 tipo IgM/IgG se ha invocado como un precedente para el desarrollo del sarcoma de Kaposi en individuos con un sistema inmunológico comprometido (24). El hecho de que el HHV8 y el sarcoma de Kaposi tengan una incidencia marcadamente más elevada en individuos homosexuales VIH+ y no se observe en hemofílicos VIH+ sugiere altamente que la transmisión sexual es la vía esencial para el HHV8. Aun así, la detección del HHV8 en linfocitos B de sangre periférica en individuos con HIV plantea la problemática de la posibilidad de su transmisión a través de transfusiones.
Tras la identificación del HHV8 en el sarcoma de Kaposi en individuos con SIDA, pudo demostrarse la presencia de este virus en: 1-lesiones de Kaposi en individuos no infectados con VIH, ej. Kaposi endémico (25, 26); 2- en un tipo muy particular de linfoma que se origina en cavidades serosas y, por ello, designado linfoma primario de cavidades (BCBL o PEL) (27, 28) y 3- en un grupo de pacientes con enfermedad de Castleman asociada o no al VIH (28).
Tabla 4- Patología asociada a HHV8
Sarcoma de Kaposi endémico y no endémico Linfoma primario de cavidades (BCBL/PEL) Enfermedad de Castleman Otros linfomas (raro) |
El sarcoma de Kaposi es una neoplasia que afecta con mayor frecuencia a varones, especialmente inmunodeprimidos, tales como portadores de VIH o bajo inmunosupresión tras transplante de órganos. Las lesiones cutáneas es la forma más frecuente de presentación aunque puede manifestarse en forma generalizada, principalmente con adenopatías, o bien afectar mucosas u otros órganos (cavidad bucal, tracto gastrointestinal, pulmón, hígado etc.). La radioterapia o el láser permiten controlar las formas cutáneas locales mientras que los cuadros generalizados requieren quimioterapia sistémica. La combinación de Interferon y Zidovudina se ha empleado, aunque los resultados son precoces.
Histológicamente, las lesiones se caracterizan por mostrar una marcada proliferación vascular con la presencia típica de células fusiformes, probablemente de origen endotelial. Existen controversias en cuanto a si estas células se originan del endotelio linfático y no se hallan relacionadas con el endotelio vascular o bien poseen características de ambos, ya que la tinción con anticuerpos presentes en los dos subtipos de células endoteliales es positiva, pero existen discrepancias en los resultados con anticuerpos que son específicos para uno u otro endotelio. En el ganglio linfático las lesiones pueden, en una fase inicial, hallarse circunscritas exclusivamente a la región capsular y/o subcapsular, lo que conlleva dificultades diagnósticas. Posteriormente el tumor se extiende -a través de los sinusoides- al resto del ganglio. Acompańando la proliferación vascular pueden observarse infiltrados de células plasmáticas, macrófagos e histiocitos.
El HHV8 se detecta en la mayoría de pacientes con sarcoma de Kaposi y SIDA. Asimismo se ha documentado en lesiones cutáneas en individuos con sarcoma de Kaposi Mediterráneo no infectados con VIH (26) y en aquellos procedentes de otras regiones como Taiwan, África y Estados Unidos (25).
Mediante estudios de PCR inverso y de ISH, el HHV8 se ha identificado en las células endoteliales y en las células fusiformes que rodean los vasos en la piel de las lesiones de sarcoma de Kaposi (29). Estudios en células mononucleares de sangre periférica en individuos con VIH demuestran la presencia de HHV8 en un 52% de los mismos, incluyendo aquéllos con lesiones de Kaposi así como otros que desarrollan las lesiones en el curso evolutivo. El HHV8 se halla preferentemente integrado en los linfocitos B (CD19+). Por el contrario el virus raramente se detecta en saliva o esputo en dichos individuos.
El BCBL o PEL es un tipo de linfoma definido por las siguientes características:
a) Se origina en cavidades serosas, no objetivándose masas tumorales.
b) Las células son morfológicamente indiferenciadas, de tamańo grande, cromatina reticular, citoplasma basófilo y, a veces, poseen características de inmunoblastos (Fig. 10).
c) El inmunofenotipo es nulo (no B no T) con pérdida marcada de la mayoría de antígenos B, incluyendo aquellos que detectan moléculas que forman parte del receptor de células B, como el CD79b, ausencia de expresión de moléculas importantes de adhesión, tales como el CD11a, CD18, ICAM-1 y del CD44 ("homing" receptor) entre otras y expresión de antígenos de activación tales como el CD23, CD25, CD38 y EMA.
d) Un genotipo B con reordenamiento de las cadenas de la Ig.
e) Una coexistencia frecuente con el VEB sin que el c-myc se halle reordenado, a diferencia del linfoma de Burkitt (28).
EL PEL se observa comúnmente en individuos con SIDA y puede asociarse al sarcoma de Kaposi. Raramente se han documentado linfomas HHV8+ que se originan en tejidos sólidos en individuos con SIDA. Principalmente éstos corresponden a linfomas primarios de sistema nervioso central y, más raramente, a aquellos que se originan en el intestino.
Se han descrito líneas celulares derivadas de pacientes con PEL que contienen HHV8 con o sin la presencia del VEB (27, 30). Estudios moleculares han demostrado que el virus se halla integrado en el núcleo de las células en forma episomal y a nivel ultraestructural pueden detectarse nucleocápsides de 110 nm en el núcleo y partículas virales completas en el citoplasma. La inoculación intraperitoneal de tales líneas celulares en ratones con inmunodeficiencia da lugar al desarrollo de linfomas sin formación tumoral, como ocurre en pacientes con PEL (30).
La enfermedad de Castleman fue descrita inicialmente en la década de los 50. Es un síndrome linfoproliferativo policlonal que afecta a una variedad de tejidos linfoides o no linfoides y, en su forma sistémica, suele acompańarse de sintomatología general. Dicho cuadro se halla íntimamente relacionado al sarcoma de Kaposi coexistiendo ambos procesos en un mismo individuo y, muy particularmente, en aquellos pacientes con evidencia de infección por VIH. Desde un punto de vista clínico, la enfermedad de Castleman se manifiesta como una masa local, habitualmente mediastínica aunque puede tener otras localizaciones, ej. pélvica, o bien ser generalizada, designada esta última enfermedad de Castleman multicéntrica. Las formas localizadas tienen buena respuesta al tratamiento quirúrgico mientras que las generalizadas precisan de tratamiento sistémico que generalmente incluye prednisolona; una respuesta clínica al análogo de las purinas clorodeoxiadenosina ha sido documentada en unos pocos casos.
Desde un punto de vista histológico se distinguen:
- La variante hialinovascular.
- La variante de células plasmáticas.
- La forma mixta, en la que no existe predominio de ninguno de estos dos componentes.
La forma más frecuente es la variante hialinovascular en la que la histología ganglionar muestra atrofia de centros germinales ("burn-out"), expansión de la zona del manto y paracortical con proliferación e hialinización vascular (Fig. 11 y 12). Pueden observarse células plasmáticas pero no son prominentes como en la variante plasmática. En esta última forma, no se evidencia atrofia de los centros germinales, la proliferación vascular no es tan prominente y la zona del manto se halla rodeada de una corona de células plasmáticas generalmente de naturaleza policlonal, aunque estudios de ADN han demostrado la presencia de clonas en algunos casos. La inmunohistoquímica muestra una trama dendrítica marcada, reactividad para el factor VIII en el endotelio vascular y la presencia de células que marcan para kappa y lambda. No se han descrito alteraciones de CD4 y CD8 en los ganglios linfáticos.
El HHV8 se detecta prácticamente en todos los casos de enfermedad de Castleman en individuos con SIDA y hasta en un 40% de enfermos sin evidencia de VIH, concomitante o no al sarcoma de Kaposi (28, 31).
La relación entre la enfermedad de Castleman y el sarcoma de Kaposi es incierta, si bien ambos poseen una serie de rasgos en común tales como proliferación vascular de los tejidos y anomalías del sistema inmunitario, por ejemplo la hipergammaglobulinemia policlonal y, menos frecuentemente, la anemia hemolítica autoinmune, se asocian a enfermedad de Castleman.
Existen controversias respecto a si el HHV8 se halla presente en tejido prostático y del tracto urogenital en individuos normales. Si bien algunos estudios han documentado su presencia mediante PCR, otros no han confirmado tales hallazgos. Se ha descrito la presencia de HHV8 en células dendríticas de médula ósea de pacientes con mieloma múltiple y se ha especulado que la infección de las mismas conllevaría a la hipersecreción de IL-6 responsable de la proliferación de células plasmáticas.
Asimismo, el HHV8 se ha identificado en lesiones cutáneas de individuos con cuadros de inmunodeficiencia no portadores de VIH y en una proporción de tumores de naturaleza vascular tales como angiosarcoma o hemangioma.
Por el contrario, el HHV8 no se ha identificado en otros procesos neoplásicos cutáneos tales como micosis fungoides, linfoma anaplásico cutáneo Ki-1+, papulosis linfomatoide o EH.
El virus de la hepatitis C (VHC) es un virus ARN de la familia flavivirus que posee un tropismo in vivo e in vitro para el hepatocito y linfocito. Es el agente causal más frecuente de la hepatitis no A no B y un factor predisponente al desarrollo de carcinoma hepatocelular.
El papel que juega el VHC en el desarrollo de linfomas es controvertido. El VHC se halla involucrado en una serie de síndromes que afectan el sistema linfoide tales como la crioglobulinemia mixta, cuadro que siendo no clonal puede abocar a linfoma. La relación del VHC con linfomas se basa en:
- Estudios serológicos que demuestran que los pacientes con linfomas poseen una prevalencia de anticuerpos anti-VHC significativamente más elevada que la población control.
- La frecuente asociación de estos linfomas en individuos seropositivos con la crioglobulinemia mixta (32, 33).
El tipo de linfoma asociado al VHC es de origen B y, generalmente, de bajo grado, principalmente inmunocitomas, linfomas MALT y de zona marginal y con menor frecuencia centrofolicular; raramente se ha descrito asociado a la EH. El genotipo VHC2ac es el que se halla preferentemente involucrado, siendo más raro el subtipo VHC1b.
A pesar de esta relación estrecha, no se ha demostrado que el VHC sea un agente causal directo en el desarrollo de estos linfomas ni tampoco su integración clonal en las células tumorales. Por consiguiente, es posible que su papel sea secundario provocando una disregulación del sistema inmunitario. Por ejemplo, se ha especulado que el VHC da lugar a una proliferación policlonal de linfocitos B que, en una fase inicial, conlleva a la producción de anticuerpos (ej., factor reumatoide) y/o crioglobulinas y que esta población sería más susceptible a adquirir cambios secundarios que darían lugar a la proliferación de una clona B y linfoma.
Éste es un tipo Beta de virus herpes que en la infancia produce un cuadro febril y exantema súbito así como otros cuadros clínicos inespecíficos de tipo vírico. Se desconoce su ciclo en el hombre pero es muy probable que, tras la infección, el virus quede acantonado en el organismo y se reactive en estados de inmunodeficiencia. EL HHV-6 se ha detectado mediante PCR en una proporción relativamente pequeńa de linfomas B en individuos inmunocompetentes y en inmunodeprimidos con SIDA así como en la EH. Su papel como agente etiológico de tales procesos linfoproliferativos es incierto, ya que se han detectado secuencias de HHV-6 en tejidos no linfomatosos en algunos de estos pacientes, principalmente en aquellos con VIH. De forma excepcional, se ha demostrado la integración clonal del HHV-6 en las células linfomatosas en un caso de linfoma de Burkitt con la t(8;14) sin evidencia de infección por el VEB (34).
Los avances en la tecnología molecular y en los campos de epidemiología y virología han permitido, a lo largo de las dos últimas décadas, establecer un lazo de unión estrecho entre la infección por determinados tipos de virus y el desarrollo de neoplasias en el hombre, fundamentalmente linfomas. Mientras que se halla plenamente demostrado el papel etiológico primario de algunos de estos virus: HTLV-I, VEB, HHV8 en el desarrollo de algunos de los tumores, el papel que pueden poseer otros, tales como VIH o el VHC probablemente sea indirecto o secundario, incrementando la susceptibilidad del individuo a adquirir otros cambios genéticos o mutaciones que aboquen a la neoplasia.
Abreviaturas
BCBL/PEL: Linfoma primario de cavidades
EH: Enfermedad de Hodgkin
HHV-6: Virus herpes-6
HHV-8: Virus herpes-8
HTLV-I Virus de la leucemia linfoma T
ISH: Hibridación in situ
KSV: Virus del sarcoma de Kaposi
IL: Interleukina
LLTA: Leucemia linfoma T del adulto
LLG: Leucemia de linfocitos granulares
NK: Natural killer
PCR: Reacción en cadena de la polimerasa
VEB: Virus de Epstein-Barr
VHC: Virus de la hepatitis C
- IARC monographs on the evaluation of carcinogenis risks to humans. Human immunodeficiency viruses and human T-cell lymphotropic viruses. Vol 67, WHO. IARCPress.
- Franchini G. (1995). Molecular mechanisms of human T-cell leukaemia lymphotropic virus type I infection. Blood, 86: 3619-3639.
- Blattner W.A. & Gallo R.C. (1994). Epidemiology of HTLV-I and HTLV-II infection. In: Adult T-cell leukaemia. Ed: Takatsuki K. Oxford University Press, pp: 45-90.
- Matutes E. & Catovsky D. (1992). Adult T-cell leukaemia lymphoma. In: Leukaemia. 2d Edition, Ed: Whittaker J.A. Blackwell Scientific Publications, London, pp: 416-433.
- Loughran T.P., Sherman M.P., Ruscetti F.W., Frey S., Coyle T., Montagna R.A., Jones B., Starkebaum G., Poiesz B.J. (1994). Prototypical HTLV-I/II infection is rare in LGL leukaemia. Leukaemia Research, 18: 423-429.
- Beral V., Peterman T.A., Berkelman R., Jaffe H. (1991). AIDS associated non Hodgkin's lymphoma. Lancet, 337: 805-809.
- Carbone A., Gloghini A., Gaidano G., Cilia A.M., Bassi P., Polito P.,et al (1995). AIDS-related Burkitt's lymphoma. Morphologic and immunophenotypic study of biopsy specimens. Am.J.Clin.Pathol, 103: 561-567.
- Raphael M., Gentilhomme O., Tulliez M., Byron P.A., Diebold J., for the French Study group of Pathology for HUman Immunodefieciency Virus-associated Tumours. (1991) Histopathological features in high grade non-Hodgkin's lymphomas in acquired immunodeficiency syndrome. Arch Pathol. Lab. Med., 115:15-20.
- Levine A.M. (1993). AIDS related malignancies: the emerging epidemic. J.Nat.Cancer Inst., 85: 1382-1397.
- Knowles D.M. (1993). Biologic aspects of AIDS-associated non-Hodgkin's lymphoma. Curr.Opin.Oncol., 5:845-851.
- Chan J.K., Sin V.C., Wong K.F., Ng C.S., Tsang W.Y., Chan C.H. et al. (1997). Non-nasal lymphoma expressing the natural killer marker CD56: a clinicopathologic study of 49 cases of an uncommon aggressive neoplasm. Blood, 89: 4501-4513.
- Nakamura S., Kato E., Koshikawa T., Yatabe Y., Nagasaka T., Ishida H. et al (1997). Clinico-pathologic study of nasal T/NK cell lymphoma among Japanese. Pathol. Int., 47:38-53.
- van de Rijn M., Bhargava V., Molina-Kirsch H., Carlos-Bregni R., Warnke R.A., Cleary M.L. et al. (1997). Extranodal head and neck lymphomas in Guatemala: high frequency of Epstein-Barr virus associated sinonasal lymphomas. Hum.Pathol, 28: 834-839.
- Chiang A.K., Chan A.C., Srivastava G., Ho F.C. (1997). Nasal T/natural killer (NK) cell lymphomas are derived from Epstein-Barr virus infected cytotoxic lymphocytes of both NK and T-cell lineage. Int.J.Cancer, 73:332-338.
- Chiang A.K., Tao Q., Srivastava G., Ho F.C. (1996). Nasal NK and T-cell lymphomas share the same type of Epstein-Barr virus latency as nasopharyngeal carcinoma and Hodgkin's disease. Int.J.Cancer, 68:285-290.
- Hart D.N.J., Baker B.W., Inglis M.J., Nimmo J.C., Starling G.C., Deacon E. et al (1992). Epstein-Barr viral DNA in acute large granular lymphocytes (natural killer) leukaemic cells. Blood, 79: 2116-2123.
- Pellenz M., Zambello R., Semenzato G., Loughran T.P. (1996). Detection of Epstein-Barr virus by PCR analyses in lymphoproliferative disease of granular lymphocytes. Leukemia and Lymphoma, 23:371-374.
- Lin A.Y., Kingma D.W., Lennette E.T., Fears T.R., Whitehouse J.M., Ambinder R.F. (1996). Epstein-Barr virus and familial Hodgkin's disease. Blood, 88: 3160-3165.
- Niedobitek G., Kremmer E., Herbst H., Whitehead L., Dawson C.W., Niedibitek E. (1997). Immunohistochemical detection of the Epstein-Barr virus encoded latent membrane protein 2A in Hodgkin's disease and infectious mononucleosis. Blood, 90: 1664-1672.
- Agagnostopoulos I., Hummel M., Finn T., Tieman M., Korbuhn P. et al. (1992). Heterogeneous Epstein-Barr virus infection patterns in peripheral T-cell lymphoma of angioimmunoblastic lymphadenopathy. Blood, 80: 1804-1812.
- Weiss L.M., Jaffe E.S., Liu X-F., Chen Y.Y., Shibata D., Medeiros J. (1992). Detection and localization of Epstein-Barr viral genomes in angioimmunoblastic lymphadenopathy and angioimmunoblastic lymphadenopathy-like lymphoma. Blood, 79: 1789-1795.
- Chang Y., Cesarman E., Pessin M.S., Lee F., Culpepper J., Knowles D.M., Moore P.S. (1994). Identification of herpes-virus like DNA sequences in AIDS-associated Kaposi's sarcoma. Science, 266:1865-1869.
- Neipel F., Albretcht J.C., Ensser A., Huang Y.Q., Li J.J., Friedman-Kien A.E., Fleckenstein B. (1997). Human herpes virus 8 encodes a homolog of interleukin-6. J.Virol., 71:839-842.
- Gao S.J., Kingsley L., Hoover D.R., Spira T.J., Rinaldo C.R., Saah A. et al (1996). Seroconversion to antibodies against Kaposi's sarcoma associated herpes-virus related latent nuclear antigens before the development of Kaposi's sarcoma. N. Engl.J.Med., 335: 233-241.
- Moore P.S. and Chang Y. (1995). Detection of herpes-virus like DNA sequences in Kaposi's sarcoma in patients with and those without HIV infection. N.Engl.J.Med, 332:1181-1185.
- Dupin N., Grandadam M., Calvez V., Gorin I., Aubin J.T., Havard S. et al (1995). Herpes virus like DNA sequences in patients with Mediterranean Kaposi's sarcoma. Lancet, 345:761-762.
- Gaidano G., Cechova K., Chang Y., Moore P.S., Knowles D.M., Dalla Favera R.(1996). Establishment of AIDS related lymphoma cell lines from lymphomatous effusions. Leukemia, 10:1237-1240.
- Gadiano G., Pastore C., Gloghini A., Volpe G., Capello D., Polito P., Vacher E. et al (1997). Human herpes virus type-8 (HHV8) in haemopoietic neoplasia. Leuk.Lymphoma, 24:257-266.
- Bosshoff C., Schulz T.F., Kennedy M.M., Graham, A.K., Fisher C.,Thomas A. et al (1995). Kaposi's sarcoma associated herpesvirus infects endothelial and spindle cells. Nat. Med., 1: 1274-1278.
- Boschoff C., Gao S-J, Healy L.E., Matthews S, Thomas A.J., Coignet L. et al (1998). Establishing a KSHV+ cell line (BCP-1) from peripheral blood and charaterizing its growth in nod/SCID mice. Blood, 91:1671-1679.
- Soulier J., Grollet L., Oksenhendler E., Cacoub P., Cazals-Hatem D., Babinet P. et al. (1995). Kaposi's sarcoma associated herpes-virus DNA sequences in multicentric Castleman's disease.Blood, 86:1276-1280.
- Mazzaro C., Zagonel V., Monfardini S., Tulissi P., Pussini E., Fanni M. et al (1996). Hepatitis C virus and non-Hodgkin's lymphomas. Brit.J.Haematol., 94:544-550.
- De Vita S., Sacco C., Sansonno D., Gloghini A., Dammacco F., Crovatto M. et al (1997). Characterization of overt B-cell lymphomas in patients with hepatitis C virus infection. Blood, 90: 776-782.
- Bandobashi K., Daibata M., Kamioka M., Tanaka Y., Kubonishi I., Taguchi H. (1997). Human herpes virus (HHV-6) positive Burkitt's lymphoma: establishment of a novel cell line infected with HHV-6. Blood, 90:1200-1207.