Comunicación
Nş 007

Comunicación |
|



Dres. Eduardo Brandán Recalde, Mónica Matsuzaki, Silvina Gutiérrez y M. Eugenia Andurno
1ş Cátedra de Patología. Hospital Nacional de Clínicas. Universidad Nacional de Córdoba. Córdoba. República Argentina.

El término de histiocitosis maligna fue utilizado por Rappaport (1) para una proliferación sistémica, progresiva e invasora de histiocitos y sus precursores morfológicamente atípicos. Muchos consideran como sinónimo a la reticulosis histiocítica medular, no obstante esta última denominación suele emplearse, además, para referirse a diferentes condiciones neoplásicas y reactivas (1). El término reticulosis histiocítica medular fue introducida en la literatura por Scott y Robb-Smith en 1939 (3), para una entidad clínico-patológica caracterizada histológicamente por la infiltración de histiocitos anormales de los senos medulares de los ganglios linfáticos y clínicamente con un comienzo repentino de la enfermedad, con fiebre, debilidad, linfadenopatía generalizada, hepatoesplenomegalia; y de manera progresiva ictericia, púrpura y citopenia severa. Sin embargo, se han descripto casos que transcurren de forma asintomática y en los que el aumento del bazo es el único hallazgo, lo que hace pensar que hay formas clínicas de evolución más crónica (3). Por otra parte, en la literatura (1), se hace mención de los denominados "linfomas histiocíticos verdaderos", recordándonos que el sistema histiocítico incluye dos grupos principales de células: las células presentadoras de antígenos (células dendríticas), y las células fagocíticas o células procesadoras de antígenos (macrófagos). El término "verdadero", alude a la distinción que debe realizarse en relación con la antigua denominación efectuada por Rappaport para linfomas con células grandes, que hoy pueden ubicarse en diferentes tipos que no corresponden en, strictu sensu, a tumores histiocíticos reales. Desde otra perspectiva, se ha descripto con el término de linfoma anaplásico de células grandes (LACG), un tipo de tumor que no puede ser ubicado en otras categorías de linfomas malignos de alto grado y que se caracterizan por células tumorales grandes, variables, que pueden incluso expresar tamańo gigante y que tiene un citoplasma abundante y usualmente múltiples nucléolos grandes o de mediano tamańo. Su patrón de crecimiento, a menudo, se corresponde con la denominada "histiocitosis maligna". Puede confundirse con metástasis de carcinoma indiferenciado o melanoma y, salvo raras excepciones se demuestra el antígeno CD30 con tinciones de inmunohistoquímica (Ki-1)(2 ).

Mujer de 25 ańos que comenzó repentinamente con un síndrome febril de 14 días de evolución, asociado a un malestar general expresado por astenia marcada, escalofríos, profusa sudoración y adenopatías generalizadas: cervicales, supraclaviculares, axilares y retroperitoneales. Demostró esplenomegalia sin evidencia de rash cutáneo. El laboratorio expresó leucocitosis (VH: 26.000/mm3), linfocitosis (VH: 49%), y plaquetopenia (VH: 35.000/mm3). Los extendidos de sangre periférica mostraron un profusión de células grandes. Una Rx. de tórax no mostró anomalías. Tres días posteriores a su ingreso hospitalario comenzó con oligoanuria, con edema y petequias en ambos miembros inferiores. Un control de laboratorio evidenció hallazgos similares a los antes expresados, a los que se agregó una anemia con 3.000.000 de glóbulos rojos y una Hb. de 10,3 g/l. Clínicamente, presentaba taquipnea, taquicardia y derrame pleural. Inicialmente se sospechó una mononucleosis infecciosa, pero con resultados serológicos negativos se descartó. Se obtuvo un biopsia de un ganglio cervical.
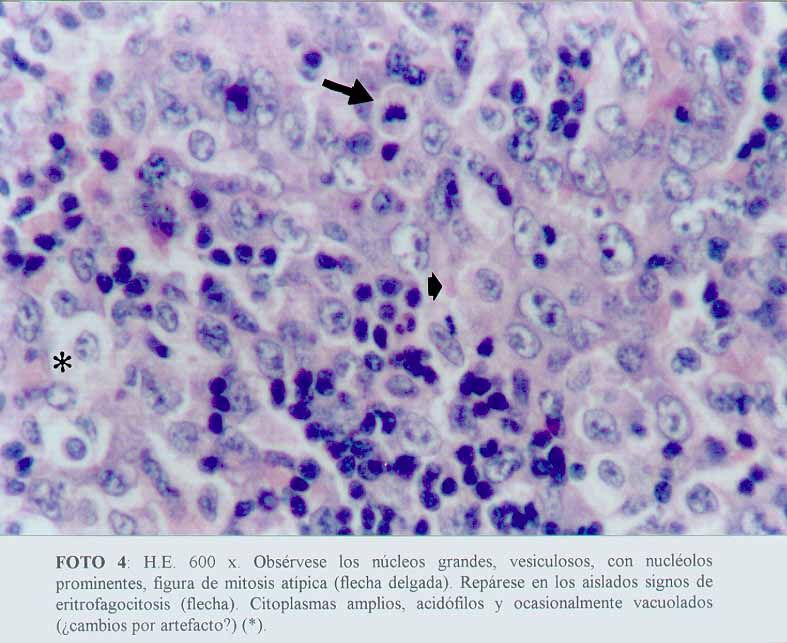
Se comprobó una proliferación de células neoplásicas, con elementos de amplios citoplasmas acidófilos, algunos claros, con núcleos grandes y pleomorfos, distribuidos de manera sincitial y con un patrón sinusoidal (microfotografía1) (microfotografía2) (microfotografía3). Se encontraron signos de eritrofagocitosis (microfotografía3) (microfotografía4). No se observó necrosis, granulomas reaccionales ni fibroplasia. La cápsula se encontraba conservada y se apreciaban cordones linfocitarios del parénquima ganglionar remanente (microfotografía2).
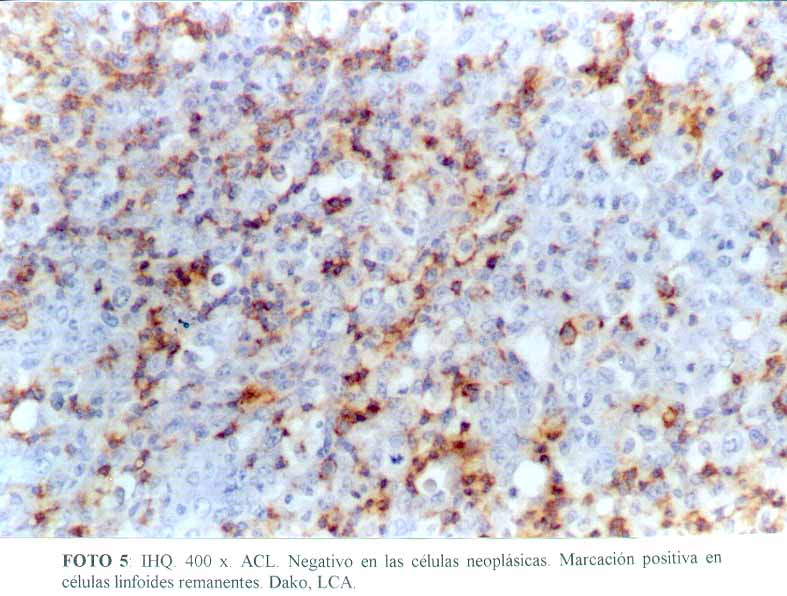
La inmunohistoquímica mostró que las células grandes, anaplásicas eran CD68 positivas (Dako KP1) (microfotografía7) (microfotografía8), expresando además, positividad para CD30 (Dako, anti CD30, Ki-1 antigen, Ber-H2) (microfotografía9) en una proporción menor de células neoplásicas, y de manera ocasional el anticuerpo contra EMA (Dako) (microfotografía10). La proteína S100 (Dako), también resultó positiva. Por el contrario, el ACL (Dako LCA) (microfotografía5), PAN B (BioGenex L26-CD 20) , CD23 (Dako CD 23 MHM6), Kappa (BioGenex LUC3), Lambda(BioGenex HP6054), PANT T (Dako, UCHL-1) (microfotografía6) y CD3 (Dako CD3) resultaron negativas. El cóctel para citoqueratinas (BioGenex AE1/AE3) dio negativo. Los extendidos de sangre periférica, previos al tratamiento, demostraron numerosas células grandes. Giemsa (microfotografía11).
Luego de dos días de un protocolo con CHOP, disminuyeron ostensiblemente. Giemsa (microfotografía12). Efectuamos el diagnóstico de un linfoma anaplásico de células grandes, de patrón sinusoidal, Ki-1 positivo, con extensa expresión de CD68, y nos planteamos la posibilidad de un histiocitosis sinusal maligna verdadera, ante la negatividad de marcadores para ACL y los marcadores disponibles para células T y B. A la paciente se le instauró una quimioterapia para LNH de alto grado (CHOP), con una rápida respuesta terapéutica y remisión completa de su cuadro clínico y analítica, recuperándose su estado general. Al momento de esta comunicación, se encuentra asintomática, realizando sus tareas habituales (médica) y sin evidencia de lesiones activas.

En los últimos ańos, se ha observado una creciente complejidad en el diagnóstico histopatológico de las neoplasias linfohemáticas, especialmente relacionada con el impacto del desarrollo tecnológico(1,2,4,6,8,11) aplicado a la observación de las entidades analizadas comúnmente por cualquier patólogo práctico. Así, desde la época de Rappaport a la actualidad, frecuentemente se han reformulado y reinterpretado diferentes cuadros, con el afán de lograr aislar entidades anatomoclínicas que sinteticen un conocimiento más acabado de su naturaleza, histogénesis, presentaciones epidemiológicas y agentes etiológicos asociados(1,4); conjunto de factores, que permitirían una proyección hacia una mejor comprensión del pronóstico y manejo terapéutico de estos tumores.
A modo de ejemplo, en el caso que comunicamos, se nos presentó como problema de diagnóstico diferencial, por la forma de presentación clínica y los hallazgos morfológicos, aunados a las expresiones de las inmunomarcaciones obtenidas, con los anticuerpos disponibles en nuestro medio, entre una histiocitosis maligna sinusal y un linfoma anaplásico con células grandes de patrón sinusoidal; ya que, resultó negativo para el ACL, como también, para rastrear un linaje B o T por inmunohistoquímica. En esta dirección, encontramos además, que el tumor fue CD30 positivo, con una marcación ocasional para EMA, y una notable expresividad para CD68 y proteína S100. Si bien nuestro panel de inmunomarcación resultó acotado, existen abundantes datos en la literatura (1,2), los cuales expresan, que aún con mayor cantidad de anticuerpos, hay circunstancias en las cuales, tampoco es posible con claridad, aislar de manera absoluta un linfoma histiocítico verdadero, de otros linfomas con células grandes, incluso el del tipo anaplásico con células grandes y, que sometidos al análisis de su genotipo, expresan el rearreglo de genes, tanto para el receptor de células T, como para el de inmunoglobulinas. En este sentido, las interpretaciones también varían (1,5,7,9,10), ya que mientras algunos piensan que estos casos serían expresiones aberrantes de linfomas histiocíticos, otros creen que con PCR se determinaría el verdadero linaje de la neoplasia, resultando la mayor parte de los casos linfomas con células grandes de tipo T.
No obstante, nuestra opinión al respecto, es preguntarnos: ¿porqué en otros campos de la patología tumoral se aceptan las diferenciaciones divergentes de una neoplasia y no en los linfomas?. De este modo, se interpela el concepto clásico de histogénesis, en el sentido que es un término que trata de definir el origen de una neoplasia desde un tipo celular, cuando en realidad lo que observa a menudo un patólogo, es un tumor que expresa distintas características morfológicas, inmunohistoquímicas y genéticas ya establecidas y por ende, lo que se somete a análisis es un fenómeno ya concretado y en progresión.
Por lo tanto, según nuestra opinión, resulta más pertinente referirse al fenómeno como fenotipoquinesis, inmunofenotipoquinesis o genotipoquinesis, haciendo alusión a que el término quinesis, del griego, significa movimiento; y por ello, el interrogante ¿de qué célula se origina?, se desvanece para dar lugar a la pregunta ¿qué está haciendo el tumor?. La respuesta puede resultar que la neoplasia esté manifestando diferenciaciones divergentes, las cuales pueden expresarse en su morfología, en sus rasgos inmunológicos o en su genotipo, sin que ninguna de ellas, per se, definan con precisión características que permitan clasificarla de una forma absolutamente acabada. Así, en nuestro caso, creemos, que mientras los expertos prosiguen sus discusiones, desde una óptica de practicidad, es mejor considerar al caso comunicado como un linfoma anaplásico con células grandes, CD30 positivo, de patrón sinusoidal, con inmunofenotipo histiocítico y de genotipo no determinado.
El futuro podrá proveernos de mayores complejidades y, por lo tanto, de notables
confusiones en el mundo (a semejanza de las opiniones del Dr. Bernard Ackerman) de la
"linfohematopatobabel".
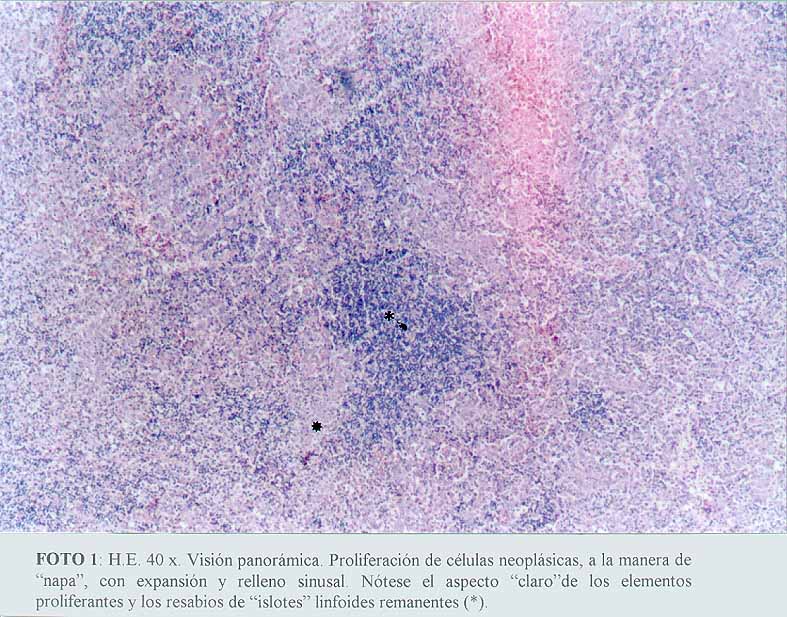 (microfotografía1)
(microfotografía1) 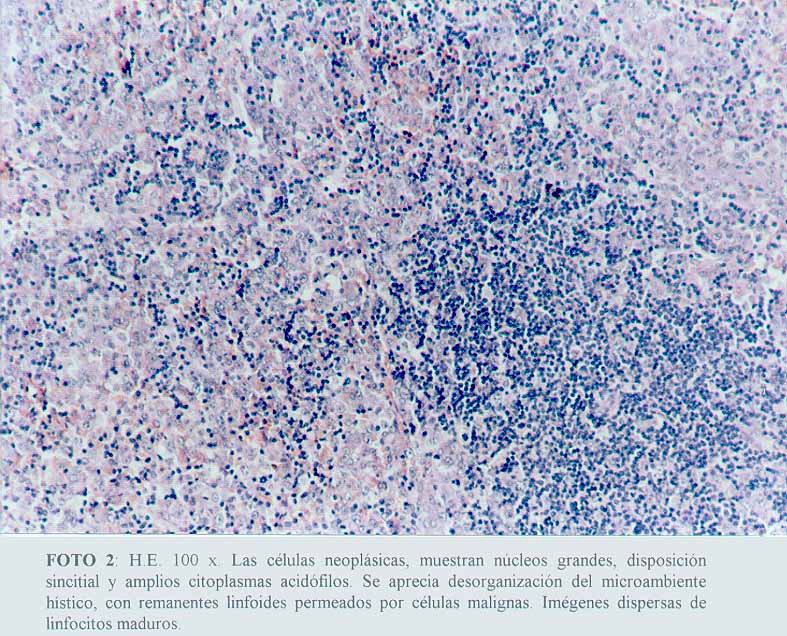 (microfotografía2)
(microfotografía2)
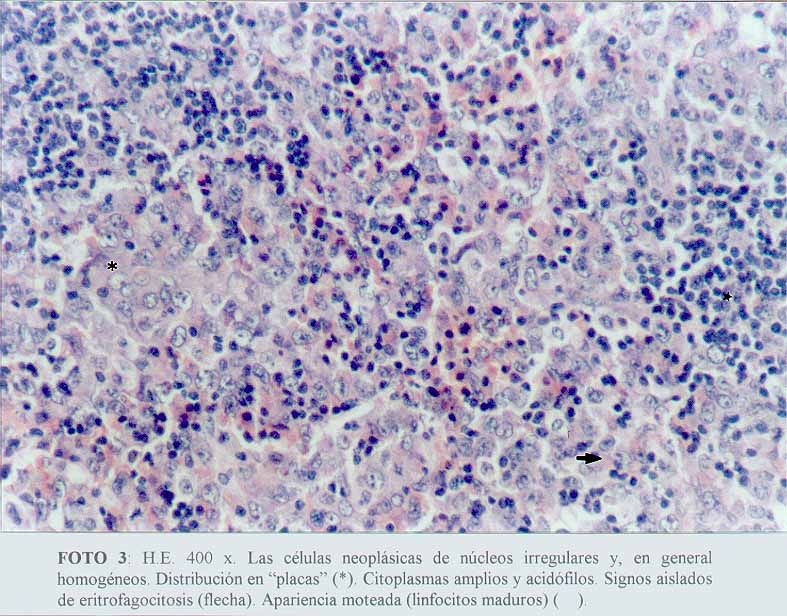 (microfotografía3)
(microfotografía3) 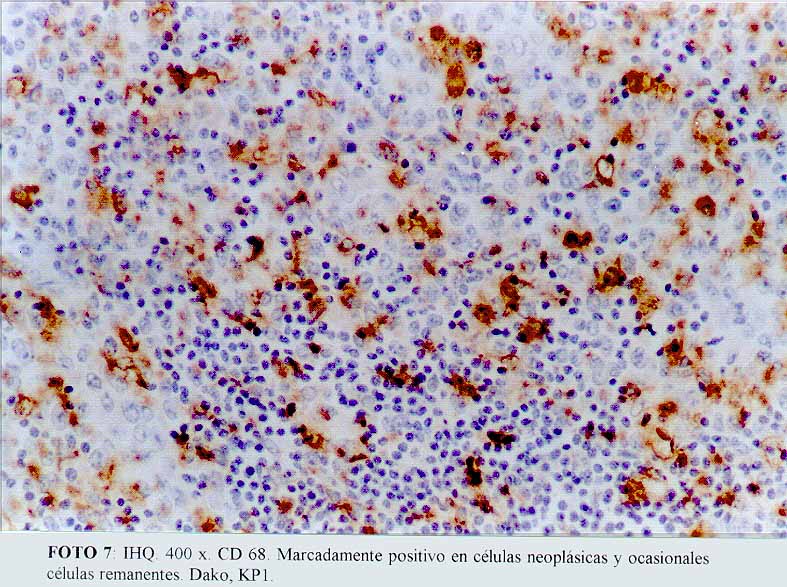 (microfotografía7)
(microfotografía7)
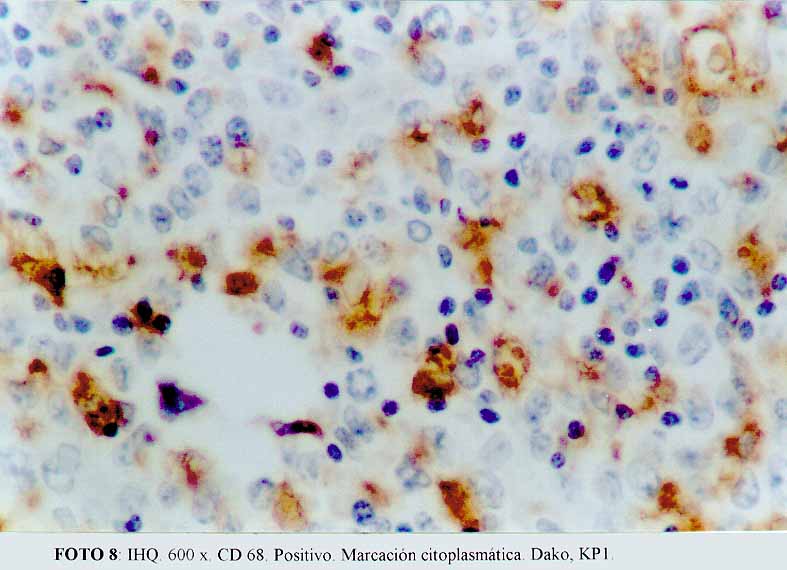 (microfotografía8)
(microfotografía8)  (microfotografía9)
(microfotografía9)
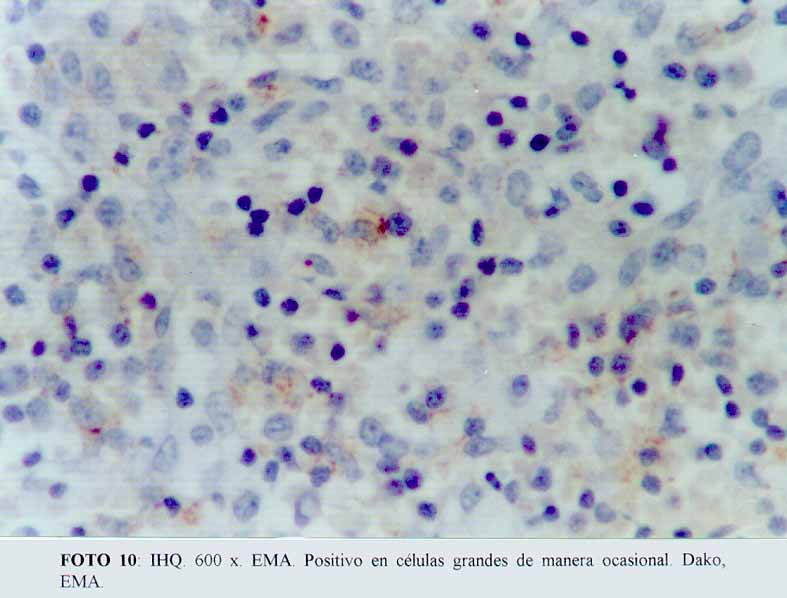 (microfotografía10)
(microfotografía10) 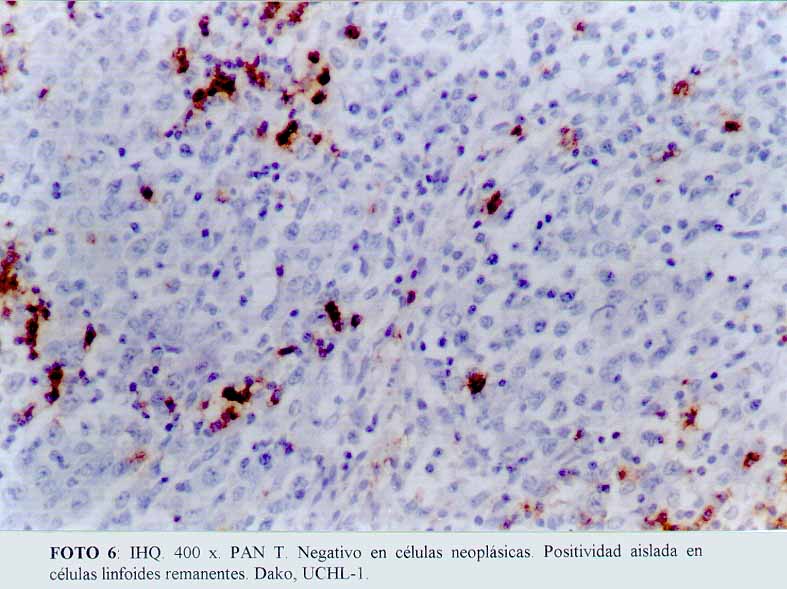 (microfotografía6)
(microfotografía6)
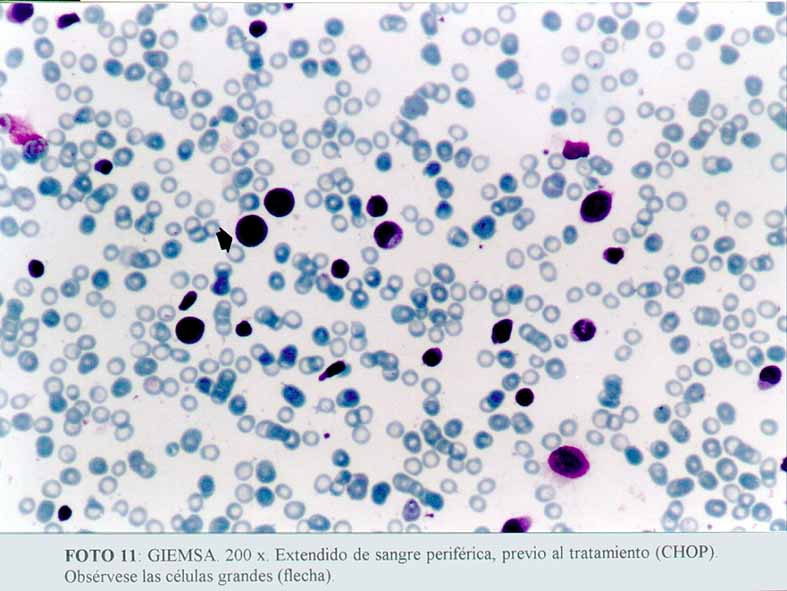 (microfotografía11)
(microfotografía11) 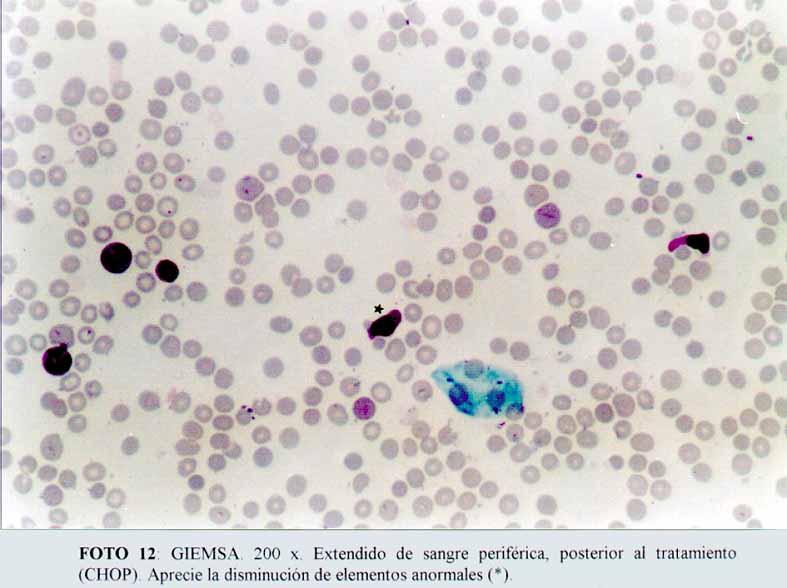 (microfotografía12)
(microfotografía12)
![]()
1-Warnke, R.A.; Weiss, L.M.; Chan, J.K.C.; Cleary, M.L.; Dorfman, R.F.: Tumors of the Lymph Nodes and Spleen. Atlas of Tumor Pathology. Third Series. Fascicle 14.Pag. 366-384. Armed Forces Institute of Pathology. Washington, D.C. 1995.
2- Lennert, K.; Feller, A.C.: Histopathology of Non-Hodgkin’s Lymphomas. 2nd Edition. Springer-Verlag. 1992.
3- Rivas, C.; Oliva, H.: Linfomas No-Hodgkin. Salvat. Pag.159-173. 1980.
4- Jaffe, E.S.; Willman, C.L.: Lymphoma and Leukemia: Integration of Morphology and Biologic Markers of Diagnosis. Long Course in Hematopathology, USCAP. March, 1998. Boston. U.S.A.
5- Turner, M.L.; Gilmore, H.N.; McLaren, K.M.; Langlands, K.; Craig, J.I.; Parker, A.C.: Regressing atypical histiocytosis: report of two cases with progression to high grade T-cell non-Hodgkin’s lymphoma. Hematol. Pathol. 7(1): 33-47. 1993.
6- Nezelof, C.: The 5q35bp chromosomal abnormality characterizes certain CD30 positive anaplastic large cell lymphoma offering a new definition of malignant histiocytosis in childhood. Nouv.Rev.Fr.Hematol. 35 (5): 463-467. 1993.
7- Mizuguchi, T.; Kohno, H.; Takishita, M.; Kosaka, M.: Malignant histiocytosis with T cell receptor and immunoglobulin gene rearrangements. Rinsho Ketsueki Dec.33 (12): 1839-1844. 1992.
8- Chang, S. C.; Wang, CH.; Su, I.J.; Chen, Y.C.; Shen, M.C.: Hematophagic histiocytosis: a clinicopathologic analysis of 23 cases with special reference of the association with peripherical T-cell lymphoma. J. Formos. Med. Assoc. May. 93 (5): 421-428. 1994.
9- Tsutsumi, Y.; Nakamura, M.; Machimura, T.: CD30 positive T cell lymphoma of the intestine, complicating ulcerative colitis. Pathol. Int. May. 46 (5): 384-388. 1996.
10- Bucsky, P.; Favara, B. Feller, A.C.; Nezelof, C.; Radzun, H.J.; Schlegelberger, B.; Janka-Schaub, G.: Malignant histiocytosis and large cell anaplastic (Ki-1) lymphoma in childhood: guidelines of differential diagnosis-report of the histiocyte society. Med.Pediatr. Oncol. 22 (3): 200-203 1994.
11- Egler, R.N.; Shmitz, L.; Sonneveld, P.; Mannival, C.; Nesbit, M.E.: Malignant histicytosis: a reassessment of cases formely classified as histiocytic neoplasms and review of the literature. Med. Pedriatr. Oncol. Jul. 25 (1): 1-7. 1995.

El Hospital Nacional de Clínicas, es el Hospital Escuela de la Facultad de Ciencias Médicas de Córdoba. Fue inaugurado el 24 de mayo de 1913, bajo la dirección del Prof. Dr. Pedro Vella, catedrático titular de Clínica Quirúrgica. La figura seńera del Hospital, padre de ilustres médicos que continúan enriqueciendo al país con su saber, fue reconocida por el Poder Ejecutivo Nacional, quién por Decreto Nro 1472/96, lo declaró Monumento Histórico, para continuar con el liderazgo de su accionar entrelazando históricamente el presente con el pasado.

Material
digitalizado y compaginado en laboratorios de  Roberto Alejandro Cabanillas Acerbi
C.E.O. INEG_Group@cordoba.com.ar
Roberto Alejandro Cabanillas Acerbi
C.E.O. INEG_Group@cordoba.com.ar
{kind=link}
{kind=link}