|
Discusión
fiogf49gjkf0d
El tejido gastrointestinal es asiento de abundante tejido linfoide y sobre todo linfocitos asociados a mucosas o MALT ( Burkard y cols 2000). Los compartimentos linfoides MALT del tubo digestivo son las placas de Peyer ( Foto 1 y Foto 2), similares a los centros germinales de los ganglios linfáticos, y por tanto predominantemente B, los linfocitos intraepiteliales de fenotipo T (CD3, CD8 y CD25 positivos y de poblaciones αβ y γδ ), los linfocitos de la lámina propia sobre todo T maduros ( CD3, CD8 y CD25 positivos) y algunos de fenotipo B diferenciados, además de macrófagos y células reticulares dendríticas y los ganglios mesentéricos (Burkard y cols 2000). En órganos dónde no existen placas de Peyer, como en el estómago, y que sin embargo, es el lugar más frecuente de asiento de linfomas de fenotipo B, de tipo MALT, surgen fenómenos de formación de estructuras como las placas de Peyer, a partir de infecciones crónicas de repetición, enfermedades autoinmunes o patología infecciosa, esta última en relación sobre todo con el Helicobacter Pylori. ( Esquema 1) ( Farinha y col 2005) Aunque la mayoría de los linfomas surgen en los ganglios linfáticos y luego se diseminan, hay una gran proporción de linfomas que nacen en compartimentos linfoides extranodales ( Burkard y cols 2000). El tracto gastrointestinal es la localización más frecuente siendo la frecuencia del 4 al 20% de los linfomas no-Hodgkin ( Ferry y cols 2004) en algunos estudios mientras que en otras series su frecuencia está descrita entre el 11 y el 34% ( Damaj y cols 2003). El tipo de linfomas es el mismo que podemos encontrar en los ganglios linfáticos. Actualmente se sigue la clasificación de la OMS ( tabla 2) aunque a partir de ella se han realizado clasificaciones de los linfomas de tipo gastrointestinal primario atendiendo a su fenotipo y frecuencia, apareciendo el concepto de casos raros misceláneos para los muy poco frecuentes ( Tabla 3) ( Ferry 2004). El orden de frecuencia de localización de los linfomas gastrointestinales en los países occidentales, es el siguiente: estomago, intestino delgado, colon y recto ( Koch y cols 2001). Estos datos coinciden con nuestros hallazgos. El recto es la localización más rara, siendo en nuestra serie de un 1,6%, que coincide con alguna serie ( Kohno y cols 2003). La media de edad de nuestra serie fué de un 66,57 años, que coincide con otras series donde se aproxima a los 60 años, afectando por igual a hombres y mujeres, mientras que en la nuestra afectan mas a mujeres con un 60,3% ( Yoon y cols 2004). En el Mediterráneo y medio oriente el intestino delgado es el más frecuente. Enfermedades inmunosupresoras como el SIDA, enfermedad inflamatoria intestinal, enfermedad celiaca e infección por H. Pylori son factores que aumentan la frecuencia ( Jaffe y cols 2001, Yoon y cols 2004). En nuestra serie hay un caso en el estomago asociado a SIDA y otro a enteropatía en el intestino delgado. Los linfomas de fenotipo B son los más frecuentes ( Damaj y cols 2003). Los localizados en el estómago suponen aproximadamente la mitad de los mismos, y en esta localización los MALT son los más frecuentes ( Damaj y cols 2003). Para clasificar los linfomas detectados en nuestra revisión seguimos la clasificación de la OMS y la de Ferry ( Tabla 2 y Tabla 3) ( Jaffe y cols 2001, Ferry 2004) . La localización según la frecuencia en nuestros casos ( Tabla 1) coincide con la frecuencia descrita en el capítulo de Ferry. Además detectamos dos linfomas linfoplasmacíticos en localización rectal. El grado de los linfomas es muy importante para el pronóstico y tratamiento (Yoon y cols 2004). El grupo Europeo de estudio del linfoma gastrointestinal, considerando las características histopatológicas de los linfomas extranodales, propuso una forma de estadiar este tipo de tumores (Paris staging system) basándose en la profundidad de la invasión tumoral, la extensión a ganglios linfáticos y a la diseminación específica de estos tumores, teniendo en cuenta también la afectación de la médula ósea (Ruskoné-Fourmestraux y cols 2003).
Los linfomas MALT, que crecen en zonas de mucosas/epitelios, representan un 8% de todos los linfomas no-Hodgkin y en el estomago son los más frecuentes ( Aviles y cols 2005). En nuestra serie también fueron los más frecuentes. Se ha implicado en su patogénesis la infección por Helicobacter Pylori, aunque en un 5-10 % no hay evidencia de ella ( Farinha y cols 2005), frecuencia similar a la de nuestra revisión, que es del 12,7% de casos en los linfomas MALT gástricos. El diagnóstico del linfoma MALT está basado en la histología y en segundo lugar en la evidencia de monoclonalidad ( Franco y cols 2005). Después de su erradicación el linfoma de bajo grado regresa en el 60-80% de los casos ( Franco y cols 2005). La presencia de nidos sólidos de células grandes no implica el término Linfoma MALT de alto grado, que debe ser desechado, sino el diagnóstico de linfoma de células grande con componente MALT acompañante ya que implica peor pronóstico ( Jaffe y cols 2001). Después del estudio de esta clase de tumores, con morfología o no de linfoma con los métodos histológicos convencionales, vimos la dificultad de clasificar los distintos tipos con la gran variedad de marcadores inmunohistoquímicos existentes. Consecuentemente decidimos realizar una serie de algoritmos y esquemas encaminados a facilitar el diagnostico con los medios básicos de un laboratorio de patología general ( Esquema 2, Esquema 3, Esquema 4 y Esquema 5).
Con respecto a los LINFOMAS DE FENOTIPO B: De todos los linfomas que se reflejan en las clasificaciones anteriormente citadas, en nuestra serie hemos detectado los siguientes tipos de linfomas primarios gastrointestinales:
- LINFOMA LINFOPLASMACITICO
Los dos únicos casos de nuestra serie estaban localizados en el recto.
Es un linfoma de fenotipo B, de células pequeñas, a veces asociado a macroglobulinemia de Waldenstrom. Morfológicamente son linfocitos plasmocitoides pequeños y células plasmáticas dispersas que suelen segregar una proteína monoclonal que puede detectarse en el suero. ( Foto 3 y Figura 4) Se excluyen de éste algunas variantes con células plasmáticas o plasmocitoides de otros linfomas ( Jaffe y cols 2001). Es raro (1,5% de linfomas nodales) y ocurre en personas mayores, con una media de 63 años, siendo ligeramente mas frecuente en varones. Los casos primitivos extranodales son excepcionales y suelen ser reflejo de extensión a estos órganos desde ganglios y médula ósea. Algunos casos han sido asociados con infecciones por virus de hepatitis C. Puede producir un síndrome de hiperviscosidad cuando la paraproteína se expresa en suero (generalmente Ig M) ( Jaffe y cols 2001). La contrapartida normal se supone un linfocito B periférico predestinado a diferenciarse en célula plasmática tras la respuesta inmune primaria a un estímulo antigénico o bien a un linfocito B post centro germinal ( Jaffe y cols 2001).
- LINFOMA DE ZONA MARGINAL, EXTRANODAL, DE TEJIDO LINFOIDE ASOCIADO A MUCOSAS (MALT)
En nuestros casos, los linfomas MALT fueron los más frecuentes en el esófago y en el estómago, mientras que en el intestino delgado tuvieron una frecuencia de 30% y en el colon del 22%.
Los linfomas MALT comprenden entre el 7 y 8% de todos los linfomas de fenotipo B. El tracto gastrointestinal es el más frecuentemente afectado y de éste el estómago (más del 50% de todos los linfomas extranodales MALT son gástricos). La media de edad de aparición está por encima de los 60 años y son ligeramente más frecuentes en mujeres.
Los linfomas MALT ( Foto 5) surgen de linfocitos que recapitulan a los de la zona marginal de las placas de Peyer, son de fenotipo B y están muy relacionados epidemiológica mente con el H. Pylori ( Jaffe y cols 2001, Ferry 2004). Son linfomas de bajo grado de malignidad, pero pueden progresar a linfomas de células grandes (de tipo centroblástico o inmunoblástico). En este caso no deben ser diagnosticados de linfomas MALT de alto grado sino de linfoma de células grandes ( Foto 6 y Foto 7). Incluso si se ve transformación parcial a acúmulos de células grandes se debe diagnosticar de linfoma de células grandes haciendo referencia a las áreas MALT residuales ( Jaffe y cols 2001). Son tumores linfoides morfológicamente muy heterogéneos que derivan de la zona marginal perifolicular situada externamente a la zona del manto ( Esquema 1). Este compartimento, está representado por células pequeñas, de morfología monocitoide, similares a los centrocitos (centrocyte-like) pero con más citoplasma pálido con la hematoxilina-eosina ( Foto 8). Como en condiciones no tumorales es una zona dinámicamente de trasiego celular desde las áreas extrafoliculares al centro germinal y viceversa, existen además de las anteriores, células similares a centroblastos, algunos inmunoblastos y células plasmocitoides en número variable. En la contrapartida neoplásica pueden formar parte de estos linfomas, aunque generalmente en menor número y explica la heterogeneidad ya reseñada. La localización perifolicular implica una localización interfolicular en los agregados linfoides y tanto en lesiones reactivas como en las neoplásicas, cuando aquellos están asociados a mucosas, suelen infiltrar los epitelios vecinos dando lugar a lesiones linfoepiteliales ( Foto 9) que son típicas de estos procesos ( Jaffe y cols 2001). Isaacson establece dos criterios histológicos que deben cumplir: remplazamiento de las glándulas por la infiltración de células uniformes y clara evidencia de destrucción glandular ( Foto 10) ( Yoon y cols 2004). Muchas veces existe un componente celular plasmocitoide numeroso pero la secreción de paraproteína a sangre periférica es rara, con la excepción de la enfermedad inmunoproliferativa del intestino delgado, variante de linfoma MALT, dónde se detecta una cadena alfa aberrante en sangre periférica.
La zona marginal, aunque anatómicamente idéntica tanto en ganglios linfáticos como en otros tejidos linfoides extranodales, está situada en primera línea de la respuesta inmune y actúa de urgencia contra muchos antígenos al margen del sistema mayor de histocompatibilidad (HLA). Por esto está desarrollada de forma desigual según el tejido linfoide que se trate y según la localización de éste. Así, en situaciones no tumorales, no se detecta morfológicamente en los ganglios linfáticos, es manifiesta en el bazo y está muy desarrollada en órganos como amígdalas, placas de Peyer y lugares de trasiego antigénico muy activo. Esta promiscuidad celular hace que, al contrario que sus vecinas, la zona marginal esté mal delimitada también inmunofenotípicamente. El CD5 y CD10 deben ser negativos, y la co-expresión de CD43 en un linfoma gástrico, que ocurre por encima del tercio de los casos, orienta hacia linfoma MALT ( Foto 11) ( Jaffe y cols 2001, Ferry 2004). En el estómago, son factores de mal pronóstico de los linfomas MALT la afectación de ganglios peri-gástricos (patrón de infiltración morfológico microscópico sinusal preservando los folículos linfoides), infiltración más allá de la mucosa, la localización proximal, el componente de alto grado y la presencia de translocaciones, que cuando ocurren son resistentes a la terapia para H.Pylori ( Foto 12) ( Jaffe y cols 2001).
- LINFOMA FOLICULAR
En nuestra serie encontramos un solo caso de linfoma de este tipo, localizado en el estómago.
Supone aproximadamente el 22% de los linfomas no-Hodgkin en el adulto y de ellos, el 70% de los denominados linfomas de bajo grado de malignidad. Es raro por debajo de los 20 años y la media de edad de aparición está en torno a los 59 años. Es ligeramente más frecuente en mujeres ( Jaffe y cols 2001). Los LF primitivos gastrointestinales son muy raros y suelen derivar de los primitivamente nodales que luego se extienden al aparato digestivo.
Son tumores que derivan del centro germinal de los folículos linfoides, por ello también llamados linfomas centrofoliculares, por lo que por definición son de fenotipo B, con reordenamiento clonal de gen de la IgH (fracción FR2-JH), tanto de los ganglios linfáticos como de los de otros órganos hematopoyéticos extraganglionares. El término folicular engloba dos conceptos fundamentales: los tumores crecen, sobre todo al comienzo, formando nódulos ( Foto 13) y están constituidos por células centrofoliculares que tienen un fenotipo específico ( Tabla 6) ( Jaffe y cols 2001). Desde el punto de vista morfológico no es un tumor homogéneo. Siempre existe una mezcla de células linfoides pequeñas (centrocitos ó linfocitos hendidos) y grandes (centroblastos), imitando las células de los centros germinales normales. Según el número de centroblastos presentes, tanto la REAL como la OMS en sus clasificaciones han dividido en tres grados a los LF.
Arquitecturalmente existen tumores mixtos, foliculares y difusos. Si el patrón folicular en superior al 75%, son linfomas foliculares. Si las áreas foliculares suponen menos del 75% y el resto son difusas los linfomas son foliculares y difusos (mixtos) y cuando menos del 25% son foliculares, el linfoma es difuso.
Tanto el patrón de predominio celular como el arquitectural tienen implicaciones pronósticas, siendo de peor pronóstico los de mayor grado y más agresivos los difusos que los foliculares.
La trasformación a linfoma difuso de células grandes ocurre entre un 25-35% de los linfomas foliculares y entonces la evolución es muy rápida y suele aparecer resistencia al tratamiento, con lo que el pronóstico es muy malo.
El manto folicular no participa en la neoplasia así como tampoco la zona marginal monocitoide, aunque ésta última en ocasiones puede ser hiperplásica, rodeando los folículos neoplásicos, dato éste que parece que ensombrece el pronóstico. La presencia de células plasmocitoides es excepcional ( Jaffe y cols 2001).
- LINFOMA DEL MANTO
Aunque nosotros no hemos tenido ningún caso de este tipo, hemos querido describirlo porque existen casos muy característicos en el sistema gastrointestinal.
Supone entre el 3 y el 10% de los linfomas no-Hodgkin, con una edad media de aparición de 60 años. Más frecuente en hombres, sobre todo es nodal aunque el lugar extranodal más frecuentemente afectado es el tracto gastrointestinal (30%) y después el anillo de Waldeyer ( Jaffe y cols 2001). Sus características clínicas, morfológicas, inmunohistoquímicas y moleculares pueden verse en las tablas 4, 5, 6 y 7.
Deriva de las células del manto folicular, situado entre el centro germinal y la zona marginal (Esquema 1). Muestra reordenamiento clonal de gen de la IgH ( fracción FR2-JH), con la región variable no mutada, lo que supone una célula B pre-germinal, aunque existen casos aislados con mutación somática como la de los linfocitos foliculares/ post-foliculares y por tanto con genotipo similar.
Generalmente es muy monomorfo, de linfocitos de pequeño ó mediano tamaño, ligeramente hendidos (como los centrocitos). Al diagnóstico es difuso aunque puede existir un patrón vagamente nodular y también en diana (patrón zona del manto). Este último biopsiado de forma incipiente, se ve el tumor expandiendo el manto pero sin borrar todavía los centros germinales y sin fundirse con los nódulos vecinos respetando los espacios interfoliculares. Acompañando al tumor frecuentemente existen histiocitos epitelioides, en ocasiones con hemosiderina, que confieren a la lesión, cuando se observa a pequeño aumento, un patrón en cielo estrellado. Así mismo pueden verse pequeños vasos hialinizados ( Jaffe y cols 2001). No suele, como otros linfomas, evolucionar a linfomas de células grandes, pero con la evolución de la enfermedad y las recaídas puede hacerse más agresivo. Muestra entonares un incremento de la actividad mitótica, aparece pleomorfismo y agrandamiento nuclear, de forma que puede cumplir los criterios de la variante blastoide. El parámetro más importante en relación con el pronóstico es el incremento del índice mitótico (más de 10- 37,5 mitosis/ 15 cga) ( Jaffe y cols 2001). Aunque morfológicamente semeja linfomas de linfocitos pequeños ó LLC-B su pronóstico es malo, no es curable y la media de supervivencia oscila entre 3 y 5 años.
Existe una forma peculiar de infiltración del aparato digestivo denominada poliposis linfomatoide de la que la que es responsable en su inmensa mayoría, el LCM.
- LINFOMA DIFUSO DE CELULAS GRANDES DE FENOTIPO B
En nuestra serie se observaron casos en el estómago, intestino delgado y colon. Uno de ellos, en el colon, expresaba CD30, era de un trasplantado renal y estaba relacionado con el virus de Epstein-Barr. Linfomas de fenotipo B con expresión de CD30, no se consideran verdaderos linfomas anaplásicos y suelen aparecer en cualquier tipo de inmunosupresión, sobre todo en el SIDA.
Es una proliferación generalmente difusa, de células cuyo núcleo es dos veces o mas el tamaño de un linfocito normal ( Foto 14). Es un grupo heterogéneo, en la actualidad en estudio exhaustivo. Constituyen entre el 30 y el 40% de los linfomas no-Hodgkin del adulto. La media de edad es alrededor de los 70 años, aunque puede aparecer en niños. Más del 40 % son extranodales, siendo el tracto gastrointestinal, y su origen gástrico o ileo-cecal, los mas frecuentes. Es ligeramente más frecuente en varones.
Sus características clínicas, morfológicas, inmunohistoquímicas y moleculares pueden verse en las tablas 4, 5, 6 y 7. Hay que incidir que en este tipo de linfomas el índice proliferativo (Ki-67) es alto.
Existen cuatro subtipos: centroblastico (generalmente de origen folicular), inmunoblastico, rico en célula T e histiocitos y anaplásico ( Jaffe y cols 2001).
En cuanto a los LINFOMAS DE FENOTIPO T, mucho menos frecuentes,de todos los que se reflejan en las clasificaciones anteriormente citadas. Más heterogéneos morfológicamente que los de fenotipo B, muestran frecuentemente patrones polimórfos e incluyen otras células no neoplásicas, como eosinófilos, otros linfocitos no tumorales, macrófagos, etc, reflejo de la frecuente secreción de citoquinas por los linfocitos T tumorales ( Foto 15). En nuestra serie hemos detectado los siguientes tipos cuyo origen es el tracto gastrointestinal:
1. LEUCEMIA / LINFOMA T PROLINFOCITICO
En nuestra serie detectamos un solo caso, localizado en el estómago.
En el tracto gastrointestinal es excepcional. Generalmente se expresa en forma de leucemia, que es muy agresiva y la constituyen células de pequeño y mediano tamaño, prolinfocitos de fenotipo T post-tímico. Su diagnostico en órganos distintos de piel, médula ósea, hígado, bazo y sangre periférica se supone extensión de la enfermedad, aunque en una biopsia aislada, sin datos clínicos, con las característica inmunohistoquímicas que se le atribuyen no es posible descartar su origen a dicho nivel ( Jaffe y cols 2001). Sus características inmunohistoquímicas son como los linfomas T periféricos: CD45 positivo y CD3 positivo. En un 60% las células son CD4 positivo y CD8 negativo pero es distintivo de ellos la coexpresión de CD4 y CD8, hecho que ocurre en un 25% ( Jaffe y cols 2001).
- LINFOMA DE CELULAS T ASOCIADO A ENTEROPATIA
En nuestra serie detectamos un caso localizado en el intestino delgado.
Ocurre en adultos en un amplio rango de edad. Se suele asociar a enteropatías como la enfermedad celiaca. Algunos casos, sobre todo en América central y del sur han sido asociado a infección por Virus Epstein Barr.
Al momento del diagnostico suele estar limitado al abdomen. En algunos casos se disemina a ganglios linfáticos y una variedad de órganos extranodales (hígado, bazo, cerebro, corazón, medula ósea, pulmones, riñones, tiroides y otros lugares) lo cual incrementa su mortalidad ( Jaffe y cols 2001). Sus características morfológicas e inmunohistoquímicas pueden verse en tabla 8 y tabla 9.
- LINFOMA T PERIFERICO
De este tipo, en nuestra serie, detectamos dos casos, uno en estómago y el otro en el intestino delgado.
Se denominan así a linfomas de fenotipo T no específicos, que no pueden encuadrase en otras categoría bien establecidas como se refleja en la clasificación de la OMS. Suponen prácticamente la mitad de todos los linfomas T de los países occidentales. Predomina en adultos y afecta a ambos sexos por igual. Sus características morfológicas e inmunohistoquímicas pueden verse en tabla 8 y tabla 9 ( Foto 16). Existe una variante interfolicular denominada de zona T y otra rica en células epitelioides que corresponde al denominado clásicamente como linfoma de Lennert ( Jaffe y cols 2001).
- LINFOMA ANAPLASICO DE CELULAS GRANDES (T)
En nuestra serie detectamos uno en el estómago y otro en intestino delgado.
Es un linfoma T categorizado gracias a la irrupción de la inmunohistoquímica, ya que debe de expresar CD30 (Ki-1) y últimamente, en la mayoría de los casos, se ha asociado a la kinasa denominada ALK.
Sus características inmunohistoquímicas son comunes a la de los linfomas T periféricos de células grandes, y como en los de otras localizaciones deben ser positivos a CD30, ALK y EMA.
Suponen el 3 % de los linfomas no-Hodgkin del adulto, pero hasta el 30% de los linfomas en los niños. Existen dos picos de prevalencia, uno en jóvenes y otro de la tercera década en adelante. La afectación primaria extranodal sobre todo de la piel es frecuente, pero en el intestino y sistema nervioso central es rara ( Jaffe y cols 2001).
Con respecto a OTROS LINFOMAS en esta localización, pese a que están descritos en la literatura, no hemos encontrado ningún linfoma de Hodgkin primitivo gastrointestinal. Sin embargo si hemos detectado otro extremadamente raro, categorizado como LINFOMA HISTIOCÍTICO VERDADERO (Sarcoma histiocitico de la clasificación de la OMS). Es un tumor con características morfológicas e inmunohistoquímicos similares a las de los histiocitos (uno o más marcadores de macrófagos y negativos para los de células reticulares dendríticas). Sus marcadores son: CD45, CD45RO, CD68, lisozima y pueden ser positivos para Proteína S100 de forma focal. ( Foto 17 y Foto 18) No deben ser positivos para CD1a ni para marcadores de fenotipos B y T ni tampoco para fenotipo de estirpe granulocítica (Mieloperoxidasa). La conducta es muy agresiva ( Jaffe y cols 2001). Nuestro caso estaba situado en el colon como los descritos en algunas series. En cuanto a la valoración del índice proliferativo, como factor pronóstico de los linfomas gastrointestinales, sigue siendo válida la tinción inmunohistoquímica para el Ki-67 (MIB1) (Foto 19). Por otro lado en los linfomas MALT gástricos se ha demostrado que cuando la expresión de la proteína p27 disminuye o se pierde, predice un mal pronóstico y ayuda al diagnóstico de linfoma difuso de célula grande (Shirin y cols 2005).
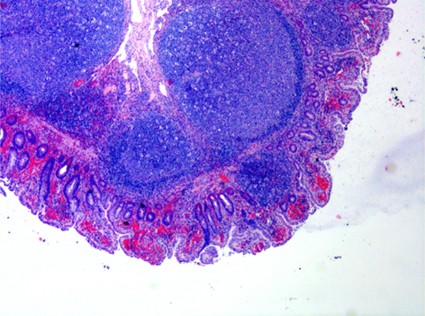 fiogf49gjkf0dFigura 1."> fiogf49gjkf0dFigura 1.">
Figura 1 - fiogf49gjkf0d Figura 1.
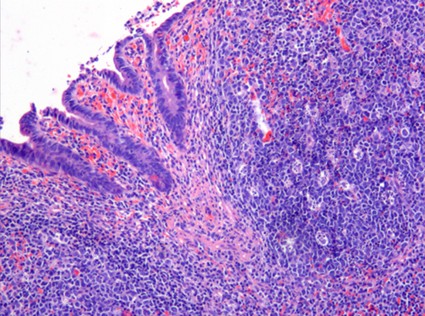 fiogf49gjkf0dFigura 2."> fiogf49gjkf0dFigura 2.">
Figura 2 - fiogf49gjkf0d Figura 2.
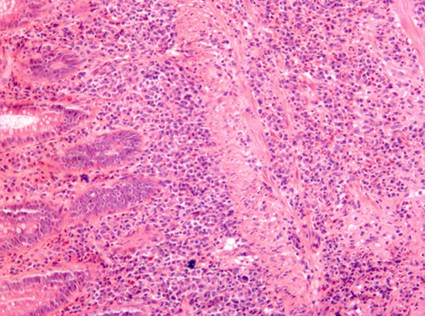 fiogf49gjkf0dFigura 3."> fiogf49gjkf0dFigura 3.">
Figura 3 - fiogf49gjkf0d Figura 3.
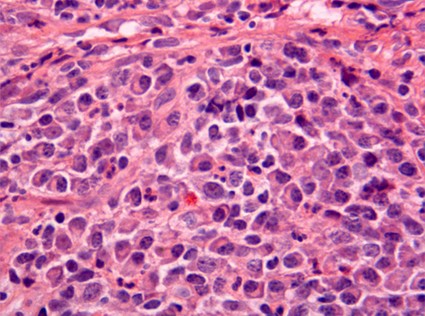 fiogf49gjkf0dFigura 4."> fiogf49gjkf0dFigura 4.">
Figura 4 - fiogf49gjkf0d Figura 4.
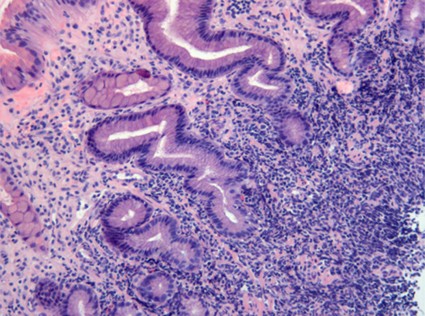 fiogf49gjkf0dFigura 5."> fiogf49gjkf0dFigura 5.">
Figura 5 - fiogf49gjkf0d Figura 5.
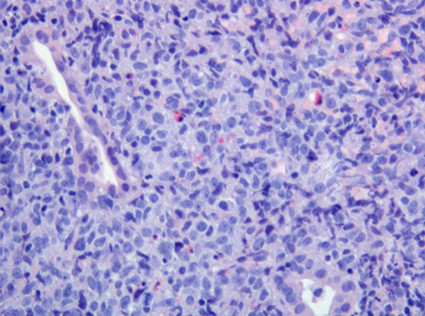 fiogf49gjkf0dFigura 6."> fiogf49gjkf0dFigura 6.">
Figura 6 - fiogf49gjkf0d Figura 6.
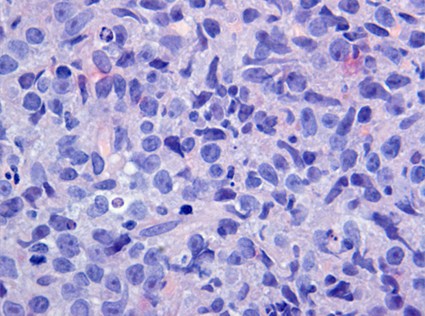 fiogf49gjkf0dFigura 7."> fiogf49gjkf0dFigura 7.">
Figura 7 - fiogf49gjkf0d Figura 7.
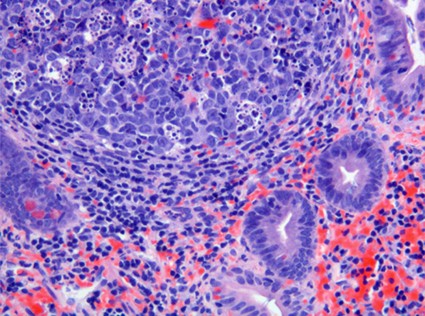 fiogf49gjkf0dFigura 8."> fiogf49gjkf0dFigura 8.">
Figura 8 - fiogf49gjkf0d Figura 8.
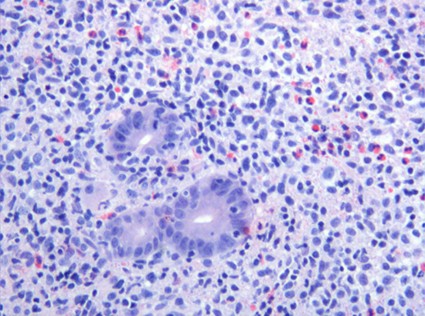 fiogf49gjkf0dFigura 9."> fiogf49gjkf0dFigura 9.">
Figura 9 - fiogf49gjkf0d Figura 9.
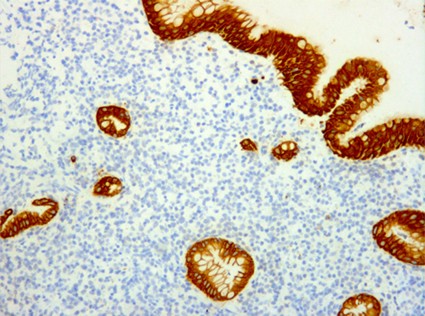 fiogf49gjkf0dFigura 10. CK"> fiogf49gjkf0dFigura 10. CK">
Figura 10 - fiogf49gjkf0d Figura 10. CK
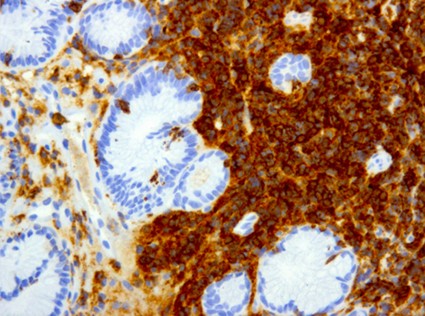 fiogf49gjkf0dFigura 11. CD43"> fiogf49gjkf0dFigura 11. CD43">
Figura 11 - fiogf49gjkf0d Figura 11. CD43
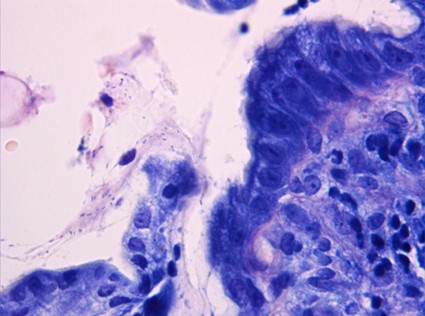 fiogf49gjkf0dFigura 12. Giemsa"> fiogf49gjkf0dFigura 12. Giemsa">
Figura 12 - fiogf49gjkf0d Figura 12. Giemsa
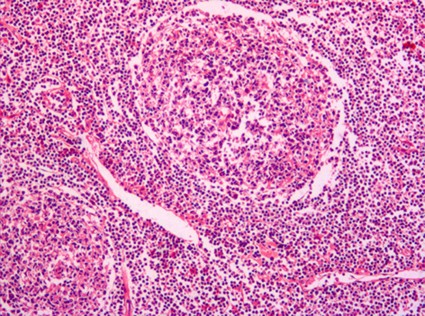 fiogf49gjkf0dFigura 13."> fiogf49gjkf0dFigura 13.">
Figura 13 - fiogf49gjkf0d Figura 13.
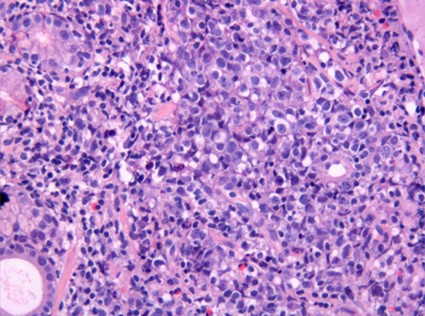 fiogf49gjkf0dFigura 14."> fiogf49gjkf0dFigura 14.">
Figura 14 - fiogf49gjkf0d Figura 14.
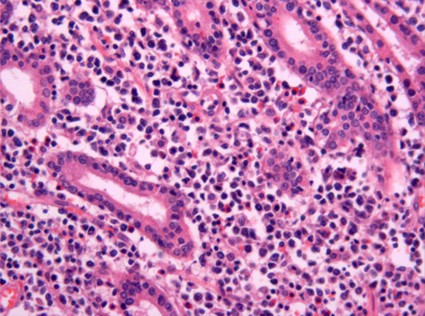 fiogf49gjkf0dFigura 15."> fiogf49gjkf0dFigura 15.">
Figura 15 - fiogf49gjkf0d Figura 15.
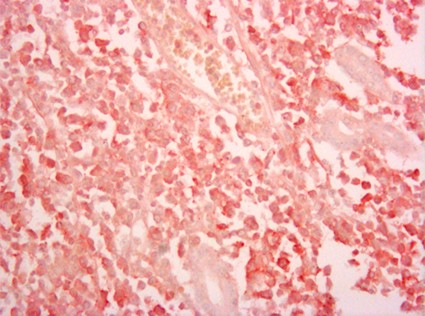 fiogf49gjkf0dFigura 16. CD3"> fiogf49gjkf0dFigura 16. CD3">
Figura 16 - fiogf49gjkf0d Figura 16. CD3
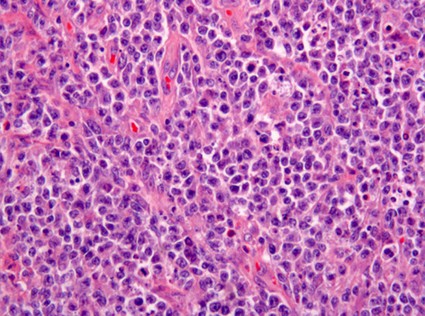 fiogf49gjkf0dFigura 17."> fiogf49gjkf0dFigura 17.">
Figura 17 - fiogf49gjkf0d Figura 17.
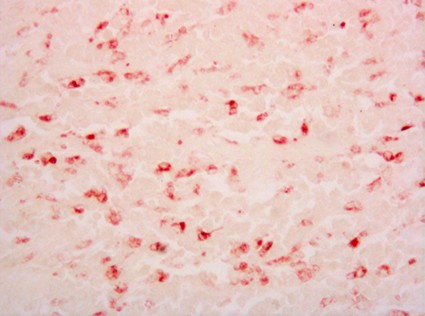 fiogf49gjkf0dFigura 18. CD68"> fiogf49gjkf0dFigura 18. CD68">
Figura 18 - fiogf49gjkf0d Figura 18. CD68
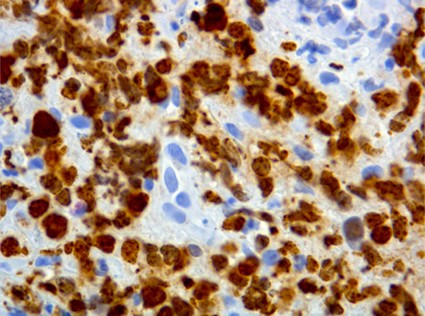 fiogf49gjkf0dFigura 19."> fiogf49gjkf0dFigura 19.">
Figura 19 - fiogf49gjkf0d Figura 19.
.jpg) fiogf49gjkf0dTabla 2. Linfomas según la clasificación de la oms detectados en nuestra revisión (OMS)"> fiogf49gjkf0dTabla 2. Linfomas según la clasificación de la oms detectados en nuestra revisión (OMS)">
tabla 2 - fiogf49gjkf0d Tabla 2. Linfomas según la clasificación de la oms detectados en nuestra revisión (OMS)
 fiogf49gjkf0dTabla 3. Linfoma primarios del tracto gastrointestinal (Ferry 2004)"> fiogf49gjkf0dTabla 3. Linfoma primarios del tracto gastrointestinal (Ferry 2004)">
tabla 3 - fiogf49gjkf0d Tabla 3. Linfoma primarios del tracto gastrointestinal (Ferry 2004)
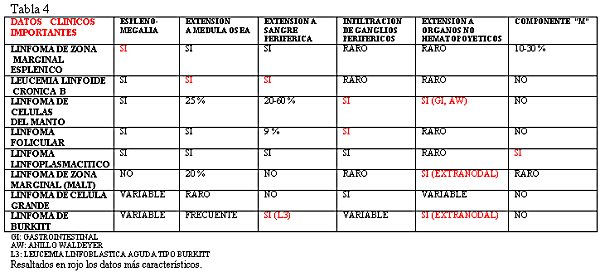 fiogf49gjkf0dTabla 4."> fiogf49gjkf0dTabla 4.">
tabla 4 - fiogf49gjkf0d Tabla 4.
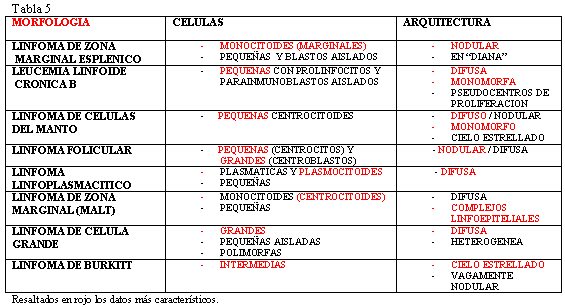 fiogf49gjkf0dTabla 5."> fiogf49gjkf0dTabla 5.">
tabla 5 - fiogf49gjkf0d Tabla 5.
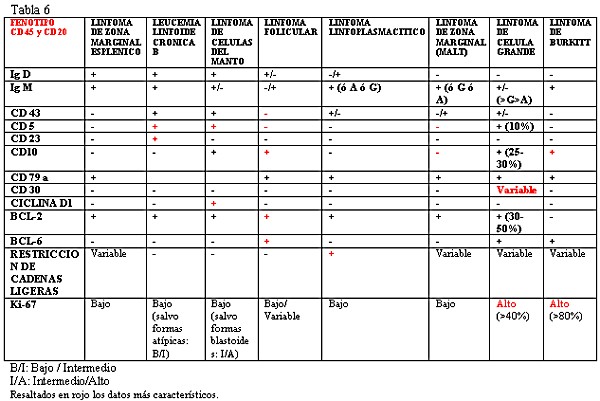 fiogf49gjkf0dTabla 6."> fiogf49gjkf0dTabla 6.">
tabla 6 - fiogf49gjkf0d Tabla 6.
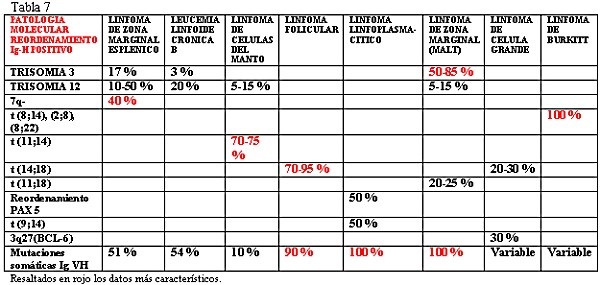 fiogf49gjkf0dTabla 7."> fiogf49gjkf0dTabla 7.">
tabla 7 - fiogf49gjkf0d Tabla 7.
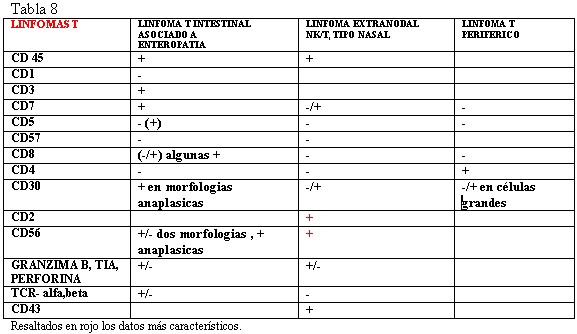 fiogf49gjkf0dTabla 8."> fiogf49gjkf0dTabla 8.">
tabla 8 - fiogf49gjkf0d Tabla 8.
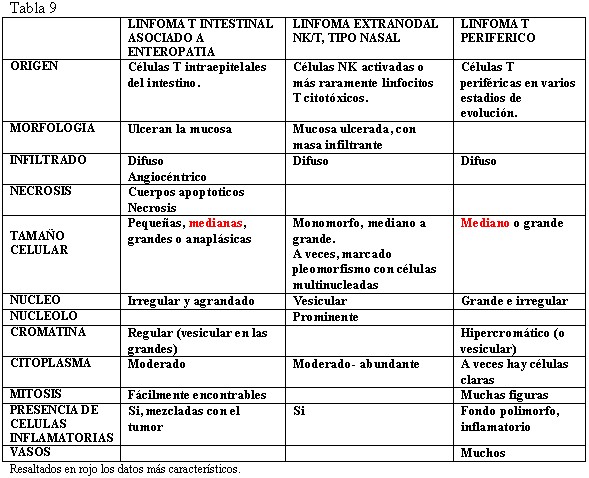 fiogf49gjkf0dTabla 9."> fiogf49gjkf0dTabla 9.">
tabla 9 - fiogf49gjkf0d Tabla 9.
|


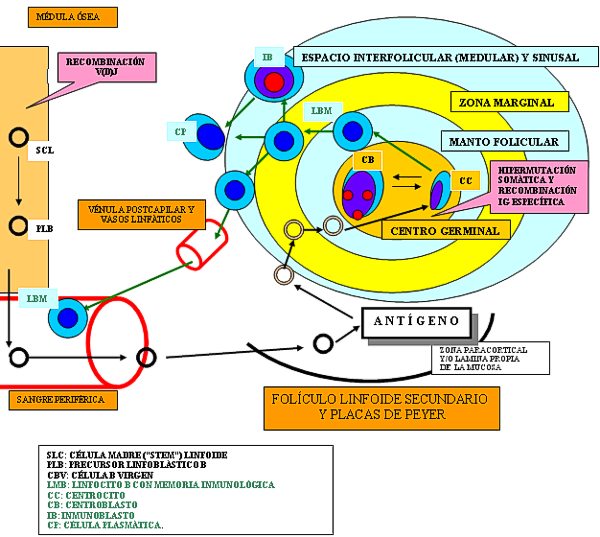 fiogf49gjkf0dEsquema 1. Origen, circulación y localización de los linfocitos B">
fiogf49gjkf0dEsquema 1. Origen, circulación y localización de los linfocitos B">
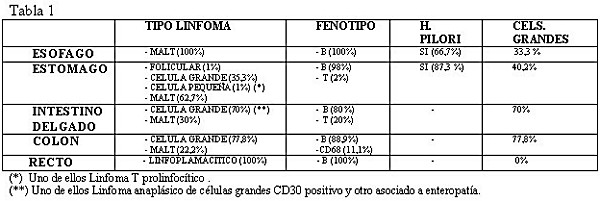 fiogf49gjkf0dResultados de nuestra casuística.">
fiogf49gjkf0dResultados de nuestra casuística.">
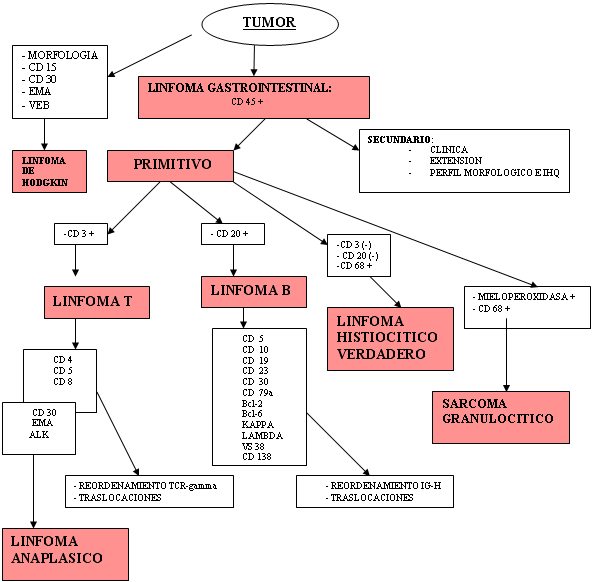 fiogf49gjkf0dEsquema 2. Algoritmo diagnóstico.">
fiogf49gjkf0dEsquema 2. Algoritmo diagnóstico.">
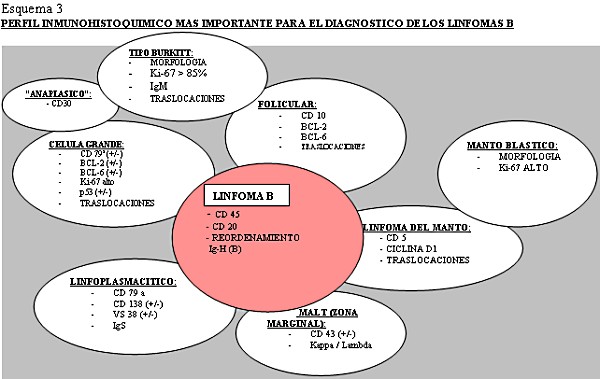 fiogf49gjkf0dEsquema 3. Perfil inmunohistoquímico mas importante para el diagnostico de los linfomas B.">
fiogf49gjkf0dEsquema 3. Perfil inmunohistoquímico mas importante para el diagnostico de los linfomas B.">
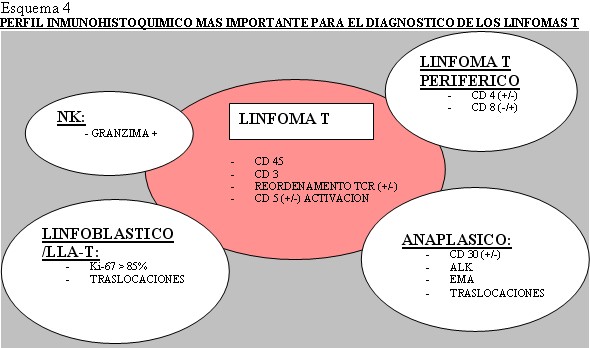 fiogf49gjkf0dEsquema 4. Perfil inmunohistoquímico mas importante para el diagnostico de los linfomas T.
">
fiogf49gjkf0dEsquema 4. Perfil inmunohistoquímico mas importante para el diagnostico de los linfomas T.
">
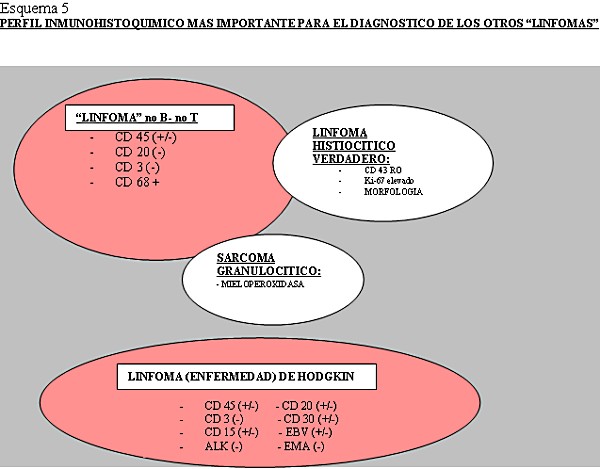 fiogf49gjkf0dEsquema 5. Perfil inmunohistoquímico más importante para el diagnostico de los otros linfomas">
fiogf49gjkf0dEsquema 5. Perfil inmunohistoquímico más importante para el diagnostico de los otros linfomas">