

|
PATOLOGÍA GÁSTRICA: Lesiones precursoras de cáncer gástrico. Revisión. JM Sanz Anquela*, A Blasco Martínez**, JM Arrinda Yeregui***, G Olmedilla Arregui* |
|
|
A escala mundial el cáncer gástrico (CG), continúa siendo la segunda causa de muerte por cáncer, superada tan solo por el cáncer de pulmón a finales del siglo XX. En la mayor parte de los casos de CG se admite un largo proceso de carcinogénesis cuyos estadíos constituyen las lesiones precursoras de CG (LPCG). El primer reconocimiento de la LPCG se remonta más de un siglo atrás, pero es en 1975 cuando Pelayo Correa presenta su hipótesis patogénica protagonizada por la secuencia: gastritis crónica atrófica metaplasia intestinal displasia carcinoma. El proceso se inicia, la mayor parte de las veces en la infancia, con la infección por Helicobacter pylori (Hp), aunque son pocas las personas infectadas que tras iniciar el proceso de LPCG llegan a desarrollar CG. El tratamiento erradicador del Hp muy probablemente retarda la progresión de la LPCG, pero no garantiza la reversibilidad del proceso en todos los casos. Se han detectado apariciones de CG tras tratamientos erradicadores y se desconoce a partir de qué estadío, o bajo qué condiciones, el efecto de la infección en el desarrollo del CG pudiera ser irreversible. La investigación de la LPCG ha transcendido poco en la práctica asistencial por varios motivos: 1.- el grado de acuerdo diagnóstico es bajo para algunas LPCG como la atrofia y la displasia. 2.- el problema del muestreo endoscópico de la mucosa gástrica sigue sin quedar resuelto para algunas lesiones como la metaplasia y displasia, a pesar de los últimos acuerdos del Sistema Sydney. 3.- entre las LPCG no se ha definido ningún marcador intermedio de carcinogénesis lo suficientemente eficaz, como para establecer protocolos consensuados de seguimiento que sean rentables. La presente revisión pretende exponer los aspectos morfológicos diferenciales de la LPCG y explorar algunas de las luces que el desarrollo de la patología molecular está proyectando sobre este grupo de lesiones. Esta revisión está dedicada a nuestro amigo y compañero Julio Torrado, recientemente fallecido y pionero en España de los estudios de carcinogénesis gástrica.INTRODUCCIÓNBajo el concepto clínico de cáncer gástrico cabrían todos los tumores malignos que se originan en el estómago; no obstante, esta revisión se va a limitar al adenocarcinoma gástrico y preferentemente al de ubicación distal, no al relacionado con el esófago de Barrett.
Numerosos autores vienen utilizando el término "precanceroso", "preneoplásico" o "premaligno" para designar determinados acontecimientos que preceden al cáncer. Esta expresión, sin embargo, tiene la connotación de "antecedente obligado", y por ello Pelayo Correa propuso el vocablo de "precursor", que implica el carácter de poder preexistir cronológicamente, pero no inevitablemente conducir a la aparición del cáncer (Correa, 1982). La terminología propuesta por Correa no era precisamente nueva, había sido utilizada más de 20 años antes, tanto por autores anglosajones (Hitchcock y col, 1957), como escandinavos (Siurala y Seppala, 1960), pero lamentablemente no caló en la literatura científica y la proposición de Correa tampoco tuvo el éxito que merecía. Consideramos nuestro deber el intentar divulgarla, al menos como homenaje al autor más relevante que durante los últimos 35 años ha sabido estar en la primera línea de investigación del cáncer gástrico (CG). La observación de que el CG puede ser precedido de cambios lesionales en la mucosa gástrica, tanto de naturaleza hiperplásica como inflamatoria, se remonta a finales del siglo XIX. Ya en 1888 Ménétrier publica 2 casos de CG, uno de ellos asociado a hipertrofia difusa de la mucosa gástrica. En 1898 Dielafoy atribuye a la inflamación y ulceración de la mucosa gástrica, descrita previamente por Cruveilhier, el riesgo de transformación en CG (Ramirez, 1994). La agresión persistente o recidivante de la mucosa, conduce a una pérdida de masa (atrofia), de celularidad principal y parietal gástrica y a un cambio en la expresión fenotípica de la celularidad de reserva, a nivel de los cuellos glandulares, representado por la metaplasia. Esta agresión puede ser inflamatoria celular (gastritis atrófica multifocal), autoinmune (anemia perniciosa), o yatrógena (cirugía gástrica). El resultado es semejante, la atrofia gástrica condiciona un estado de hipoclorhidria. El reconocimiento de la MI data de 1883, cuando Kupffer describe islotes de glándulas intestinales en la mucosa gástrica (Correa, 1982). En 1938 ya se relaciona con el CG, la gastritis atrófica que presenta metaplasia de células caliciformes. A mediados del siglo XX, Lauren por un lado y Morson por otro, describen casos de CG de parecido intestinal originados sobre mucosa gástrica con MI (Correa, 1982).
El eslabón más próximo al carcinoma es la displasia, que además de ser poco frecuente, es la lesión con menor grado de acuerdo diagnóstico a pesar de los intentos de consenso de las conferencias de Viena y Padova (Kapadia, 2003). Cuadros histológicos que patólogos occidentales interpretan como displasia, son considerados por patólogos japoneses como carcinomas (Schlemper y col, 2001), y la displasia de bajo grado se confunde fácilmente con cambios hiperplásicos Desde que Pelayo Correa y su grupo publicasen en Lancet su hipótesis sobre el modelo patogénico de carcinogénesis gástrica (Correa y col, 1975), que incluye la secuencia inflamación atrofia metaplasia displasia carcinoma, se han intentado identificar nuevos parámetros lesionales con mayor o menor riesgo de evolución a CG. Se ha definido con mayor precisión la metaplasia intestinal y se ha aclarado el misterio del factor Haenszel. Guillermo Haenszel, mediante estudios epidemiológicos de migración, postuló la existencia de un factor ambiental vinculado a las áreas de mayor riesgo de CG, que incidiendo en la temprana infancia, condicionaría el riesgo de CG en la vida adulta (Haenszel y Correa, 1975). Este aspecto clave en la cronología de la exposición medioambiental también es recogido en la hipótesis de Correa (Correa y col, 1975), apuntándose a un factor iniciador del proceso de carcinogénesis en las primeras etapas de la vida, relacionado con la ingesta. Pensando en la sal o algún mineral en el suelo o la comida, incluso se postuló que este factor iniciador debía ser común a las distintas áreas de alto riesgo de CG, a pesar de las diferencias en el entorno ambiental de las mismas (Stemmermann y col, 1977). Este factor no es otro, que la bacteria Helicobacter pylori (Hp), descubierta en la década siguiente por Marshall y Warren, y que ha revolucionado el panorama actual de la gastroenterología (Fox y Wang, 2001). La infección por Hp es de lejos, la causa más frecuente de gastritis crónica en actividad en todo el mundo y el Hp figura en la lista de carcinógenos de la clase I de la IARC (Rugge y Genta, 2005). La infección gástrica por Hp es aceptada como factor etiológico necesario, aunque no suficiente, para el desarrollo del CG, estimándose que tanto el CG como la úlcera péptica y el linfoma gástrico MALT, desaparecerían 40 años después de ser erradicada de nuestra especie la infección por Hp (Graham, 2005). Morson matiza la diferencia entre lesión (sustrato histológico) y condición (expresión clínica), repasando entre unas y otras: gastritis atrófica, úlcera gástrica, enfermedad de Ménétrier, anemia perniciosa, pólipos gástricos y displasia (Morson y col, 1980). En esta revisión no se va a entrar en detalle de las condiciones precursoras definidas por Morson, tan sólo se aludirá a las mismas a lo largo de la exposición de los parámetros que definen las lesiones precursoras. En los siguientes capítulos intentaremos presentar alguno de los logros más importantes en la comprensión de la LPCG. Afortunadamente la frecuencia del CG disminuyó drásticamente en el denominado mundo rico occidental. El logro fue inesperado y parece guardar más relación con el progreso socioeconómico que con el de la medicina. La refrigeración industrial de los alimentos y la consiguiente disminución en el uso de la sal para su conservación, han debido ser factores decisivos para este logro (Forman y Kinlen, 1991). La mayor conquista de la investigación biomédica en patología gástrica ha sido el descubrimiento del Hp. Para el control de la úlcera péptica duodenal y el manejo de las gastritis, este logro científico ha sido tan espectacular como especular. Todo lo que antes giraba en torno al pH ha pasado a girar en torno al Hp. También ha repercutido el logro en el tratamiento del linfoma gástrico tipo MALT, pero la repercusión en el control del adenocarcinoma (CG), está por llegar. A lo largo de esta exposición quedará desvelado el misterioso "enigma africano", esgrimido por aquellos que se posicionaban en contra del Hp como factor etiológico del CG. Ya no quedan investigadores que duden del papel crucial que representa la infección por Hp en el proceso de carcinogénesis gástrica. Ahora más que nunca tenemos necesidad de profundizar en el conocimiento de la LPCG, para poder seleccionar aquellos pacientes que puedan verse beneficiados por la erradicación del Hp y aquellos otros que precisarán un seguimiento estricto, con protocolos pendientes de ser consensuados.
|
||
|
|
Cuando la inflamación superficial de la mucosa gástrica, es desencadenada por Hp y exceso de sal en la dieta, representa el primer eslabón de la cadena de lesiones que en algunos pacientes puede culminar en CG (Correa, 1992). La infección por Hp adquirida en la infancia, constituye el factor iniciador más universal de carcinogénesis gástrica (Correa, 1991). La primera respuesta de la mucosa gástrica frente a la infección por Hp parece ser una gastritis aguda con infiltración epitelial de neutrófilos sin otra celularidad inflamatoria acompañante. En el 20% de los pacientes la gastritis se resolvería espontáneamente y en el resto se desarrollaría una gastritis crónica. La pangastritis por Hp es una forma frecuente de presentación en población infantil (Owen, 2003).
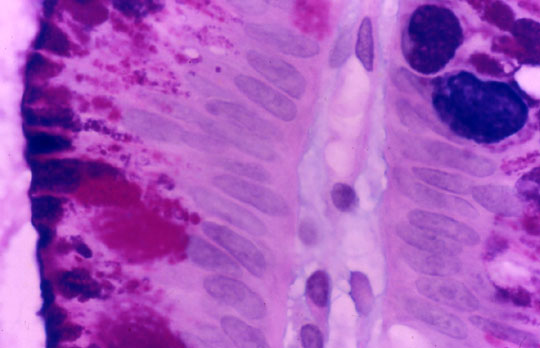
En su inicio, toda gastritis crónica comienza de forma superficial, con los infiltrados inflamatorios circunscritos a la porción más superficial de la lámina propia. Los factores iniciadores (Hp, AINES, alcohol) inducen una respuesta neutrofílica y la participación de los PMN permeando el epitelio glandular constituyen la denominada actividad inflamatoria de las gastritis activas, o en actividad. 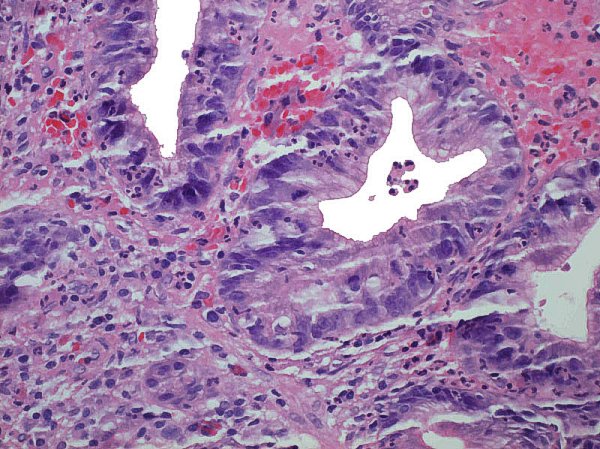 Presencia de numerosos neutrófilos (PMN), permeando el epitelio hiperplásico de invaginaciones de la superfice foveolar colonizada por Hp. ----------------------------------------------------------------------------------------------
El primer problema que nos encontramos la estudiar la mucosa gástrica es la definición de mucosa normal. La prevalencia de cambios inflamatorios es tan elevada, que el patólogo está poco acostumbrado a observar una mucosa gástrica libre de infiltrados inflamatorios. En ocasiones, discernir entre una mucosa gástrica normal y otra con cambio mínimo inflamatorio, puede resultar complicado. La mayor parte de autores están de acuerdo en que la mucosa gástrica puede alojar en su lámina propia, en condiciones de normalidad, variable cantidad de histiocitos, linfocitos y células plasmáticas, e incluso pequeños agregados linfocitarios (Owen, 1986). A diferencia de la mucosa intestinal, la mucosa gástrica normal sólo excepcionalmente incluiría folículos linfoides, los cuales constituyen un marcador de infección por Hp, también en población infantil (Carpentieri y col, 2000). Mucosa gástrica normal podemos encontrar en estómagos de niños no infectados por Hp y en los divertículos de Meckel no complicados, con mucosa gástrica heterotópica. 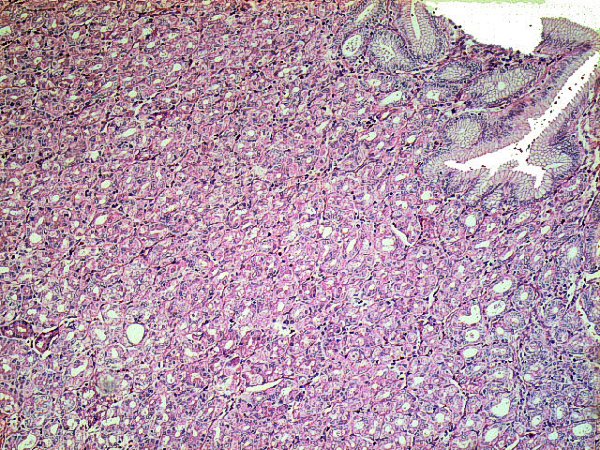 Mucosa normal tipo cuerpo gástrico, libre de infiltrados inflamatorios, procedente de una heterotopia en divertículo de Meckel. ----------------------------------------------------------------------------------------------
El segundo problema con el que nos encontramos es el de la representatividad de la muestra. La distribución de la inflamación y de otras lesiones es variable según la topografía. Para algunos autores y determinadas lesiones (MI), incluso las últimas recomendaciones del Sistema Sydney revisado (Dixon y col, 1996), serían insuficientes (El-Zimaity y Graham, 1999).
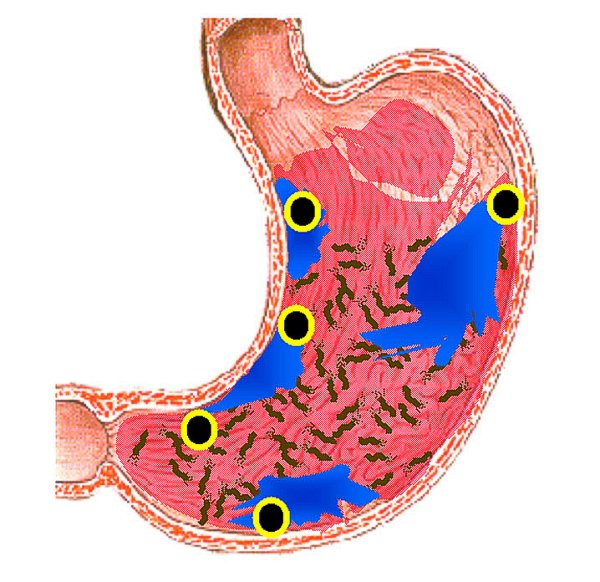 Localización topográfica de las 5 tomas biópsicas del Sistema Sydney renovado, 1994. (Esquema reproducido con permiso del Prof. RM Genta, y procedente de una de sus recientes conferencias, 2005). ----------------------------------------------------------------------------------------------
El grupo internacional de expertos reunido en Houston en 1994, para la revisión del Sistema Sydney de clasificación y gradación de las gastritis (Dixon y col, 1996), llegó a los siguientes acuerdos sobre el nivel cero de inflamación: La densidad inflamatoria medida en el seno de la lamina propia es de un máximo de 2 a 5 linfocitos, plasmáticas y macrófagos por campo de gran aumento (objetivo x40), y en la mucosa foveolar superficial de 2-3 linfocitos entre cada foveola. Sobre el epitelio superficial pueden llegar a observarse hasta 5 linfocitos por cada 100 núcleos de células epiteliales superficiales. La valoración de la densidad del componente inflamatorio debe realizarse en la mucosa alejada de posibles folículos o agregados foliculares linfoides. La incidencia repetida de los factores desencadenantes de la inflamación, condiciona los procesos recidivantes que perpetúan la respuesta inflamatoria con un doble componente celular: linfoplasmocitario y granulocítico (PMN). Ambos infiltrados inflamatorios son valorados independientemente en las escalas visuales del Sistema Sydney (Dixon y col, 1996), si bien en la última propuesta sobre gradación y estadiaje de las gastritis (Rugge y Genta, 2005), no se tiene en cuenta el tipo de infiltrado inflamatorio, sólo su intensidad y localización intragástrica. 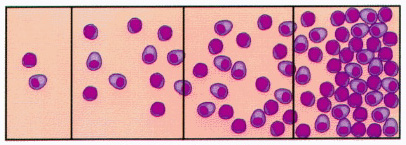 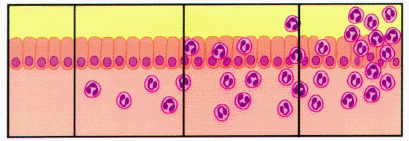 Escalas visuales de los infiltrados inflamatorios, grados 0 1 2 3 del Sistema Sydney renovado, 1994. (reproducidas con permiso del Prof. RM Genta, procedentes de una de sus recientes conferencias, 2005). 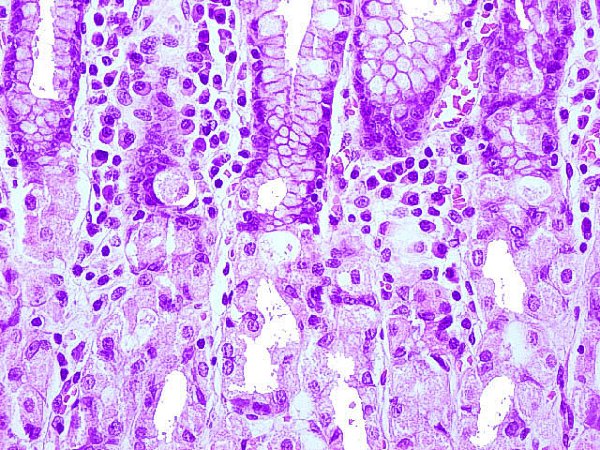 Mucosa de cuerpo gástrico con infiltrado inflamatorio grado 1 (gastritis superficial). ----------------------------------------------------------------------------------------------
El diagnóstico de las gastritis es siempre histológico, siendo el grado y tipo de inflamación su parámetro capital. Muchas son asintomáticas y muchos pacientes dispépticos carecen de gastritis. Aproximadamente el 80% de las gastroscopias se realizan por cuadros dispépticos en los que la gastritis es endoscópicamente sospechada en casi un 60% y la biopsia gástrica evidencia gastritis en más de un 70%. La discordancia entre la imagen endoscópica y los hallazgos de la histología es tan elevada, que quedaría justificada la realización de tomas biópsicas en toda gastroscopia. Las razones de esta discordancia no están claras, pero es un hecho tan evidente, que incluso la impresión endoscópica del gastroenterólogo más cualificado puede verse contradicha por un informe anatomopatológico (Owen, 2003). 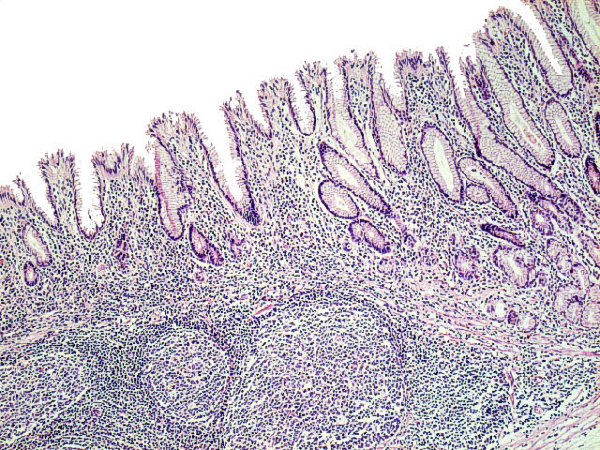 Folículos linfoides con centros claros germinales, frecuentemente hallados en las gastritis por helicobacter, en la porción profunda de la lámina propia cabalgando sobre la muscularis mucosae. ----------------------------------------------------------------------------------------------
La inflamación de la mucosa gástrica (gastritis), puede: 1.- afectar sólo al antro y no acompañarse de atrofia (gastritis difusa antral), 2.- centrarse en incisura y acompañándose de atrofia, extenderse en focos hacia cuerpo y antro (gastritis alimentaria multifocal), 3.- circunscribirse a la mucosa del cuerpo gástrico y tener un origen autoinmune, como es el caso de la gastritis corporal difusa de la anemia perniciosa (Correa, 1988). Esta clasificación de las gastritis crónicas propuesta por Pelayo Correa, fue primero ignorada en la clasificación inicial de Sydney de 1989, pero finalmente incorporada en la versión revisada del Sistema Sydney (Dixon y col, 1996).
La inflamación crónica mantenida durante largos periodos de tiempo se ha visto asociada, en general, con los procesos de carcinogénesis. No obstante, en el caso de la mucosa gástrica el desarrollo del CG no parece guardar relación directa con la inflamación sino con la atrofia gástrica secundaria a las gastritis crónicas de predominio corporal y larga evolución. La gastritis difusa antral, que carece de componente atrófico, no se ha encontrado asociada con un mayor riesgo de CG. La gastritis crónica atrófica (GCA), tanto de naturaleza autoinmune (corporal difusa), como la denominada alimentaria multifocal, sí son aceptadas como lesiones precursoras de CG (Correa, 1992). 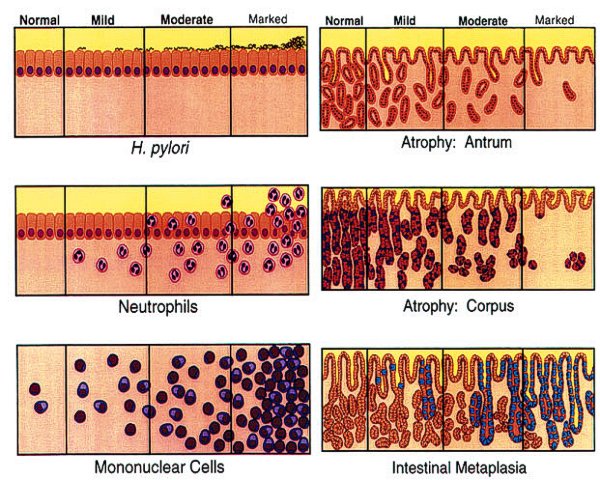 Escalas visuales de los parámetros implicados en la clasificación y gradación de las gastritis. Sistema Sydney renovado, 1994. (reproducidas con permiso del Prof. RM Genta, y procedentes de una de sus recientes conferencias, 2005) ---------------------------------------------------------------------------------------------- ¿Cómo se explica que el Hp pueda ser factor etiológico de variadas patologías, algunas de ellas contrapuestas como la úlcera péptica y el CG?. La respuesta parece estar en la genética, tanto del huésped como del alojadado. Las variaciones individuales de los genes que regulan la respuesta inflamatoria del huésped parecen ser la clave, sin olvidar variaciones en la carga citotóxica de las distintas cepas de Hp (El-Omar y col, 2001).
La inflamación es bien reconocida por el patólogo, presenta elevado acuerdo diagnóstico y su gradación en niveles de intensidad es reproducible, especialmente si se realiza con el auxilio de las escalas visuales propuestas (Dixon y col, 1996). Estas escalas ayudan menos en el caso de la atrofia, parámetro más subjetivo que precisó ser redefinido (Rugge y col, 2002). Como la topografía de la inflamación es un parámetro capital en el reconocimiento de las gastritis, dado que la inflamación de la gastritis difusa antral (no atrófica), queda limitada a la mucosa de antro, en casos de atrofia dudosa puede ser útil tener en cuenta el gradiente de inflamación cuerpo/antro. El patrón topográfico de la inflamación (intensidad de la inflamación en mucosa de cuerpo respecto a la de antro), parece ser un factor determinante para establecer el riesgo de carcinogénesis.
|
|
|
|
La mucosa gástrica atrófica ha sido
clásicamente definida como una mucosa con pérdida glandular, pero a
pesar de esta simple definición el grado de acuerdo diagnóstico para
establecer su existencia y gradación ha sido bajo (Genta, 1996),
incluso utilizando las escalas visuales propuestas por el sistema
Sydney revisado (Genta, 1998).
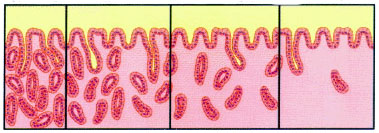 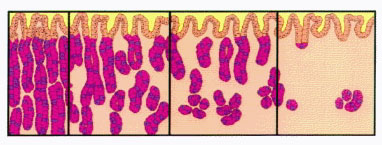 Escalas visuales del grado de atrofia en mucosa antral y de cuerpo 0 1 2 3 (normal / atrofia leve, moderada, severa) del Sistema Sydney renovado, 1994 (reproducidas con permiso del Prof. RM Genta). ----------------------------------------------------------------------------------------------
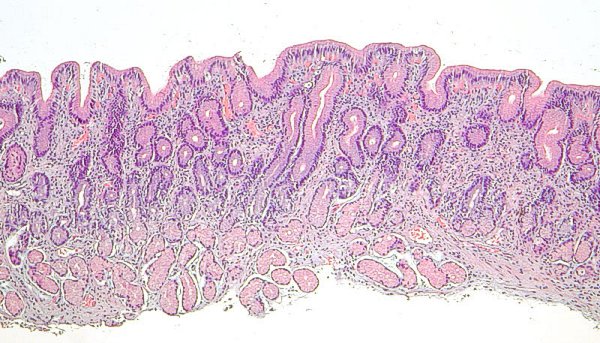 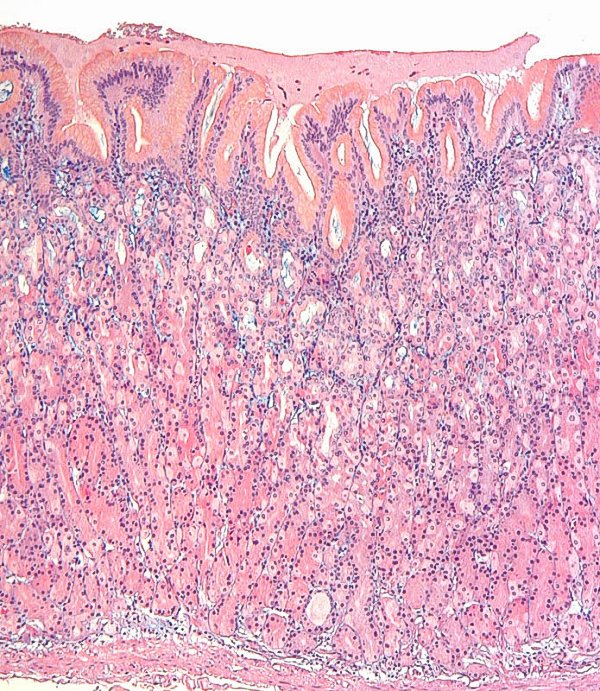 Las imágenes muestran mucosa de antro y cuerpo gástrico sin evidencia de atrofia (reproducidas con permiso del Prof. RM Genta). ----------------------------------------------------------------------------------------------
Intentando paliar las dificultades en
el reconocimiento y gradación de la atrofia gástrica, 8 de los miembros
del grupo internacional de expertos que revisaron el sistema Sydney en
1994 en Houston (Dixon y col, 1996), formaron con otros patólogos
expertos un nuevo grupo internacional que se reunió 3 veces entre 1997
y 2000, alcanzando un acuerdo de gradación de la atrofia gástrica y
redefiniéndola como una perdida de glándulas apropiadas, enlazando
así con el concepto de metaplasia (Rugge y col, 2002).
Cuando las glándulas gástricas quedan
separadas entre si, al ser desplazadas por el componente inflamatorio
que expande la lámina propia, resulta difícil decidir si la disminución
de la densidad glandular es real o aparente. Si encontramos glándulas
que no corresponden a su ubicación habitual (metaplasia), podemos
diagnosticar atrofia ya que toda transformación metaplásica que afecte
a una estructura glandular completa, representa una evidencia
inequívoca de atrofia (Rugge y col, 2002). Por el contrario, la
presencia aislada de grupos de células epiteliales intestinales en la
superficie gástrica foveolar, que no forman glándulas, representa un
fenómeno metaplásico que no implica pérdida de glándulas adecuadas
(atrofia).
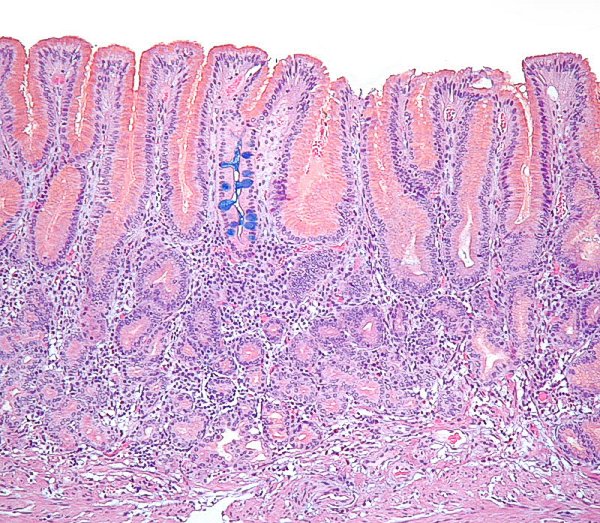 Mucosa de cuerpo con sustitución metaplásica focal del epitelio foveolar superficial por células caliciformes y enterocitos. Este hallazgo no representa una metaplasia intestinal que afecte una unidad glandular completa, por lo que no constituye marcador de atrofia (imagen reproducida con permiso del Prof. RM Genta). ----------------------------------------------------------------------------------------------
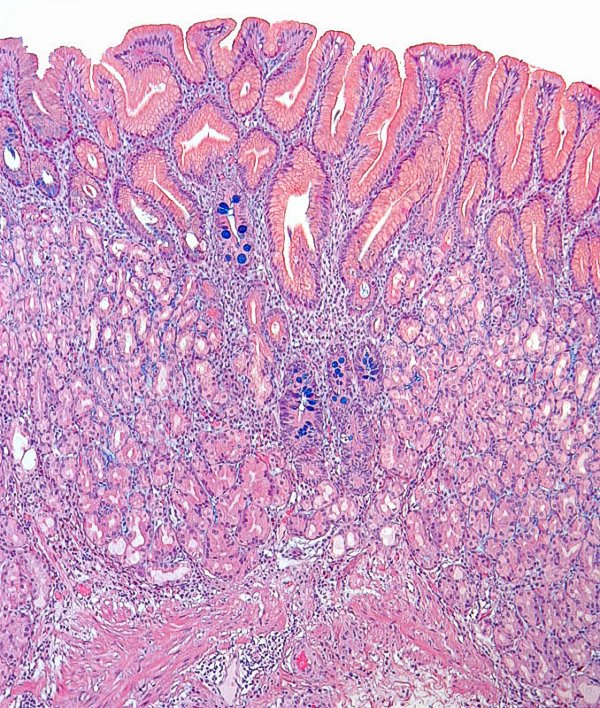 Mucosa de cuerpo con sustitución metaplásica que afecta a una unidad glandular completa y constituye marcador de atrofia, en este caso de grado leve (imagen reproducida con permiso del Prof. RM Genta). ----------------------------------------------------------------------------------------------
En la valoración de la atrofia solo
caben 3 posibilidades: ausente, indefinida y presente. La categoría de
atrofia indefinida exige una reevaluación cuando remita el componente
inflamatorio y no debe utilizarse cuando haya MI que afecte al menos a
una unidad glandular completa. La atrofia (categoría de atrofia
presente), a su vez se dividiría en metaplásica o no metaplásica, y
ambas en los 3 niveles de leve, modera y severa, representados en la
escala visual que proporciona el Sistema Sydney revisado (Dixon y
col, 1996).
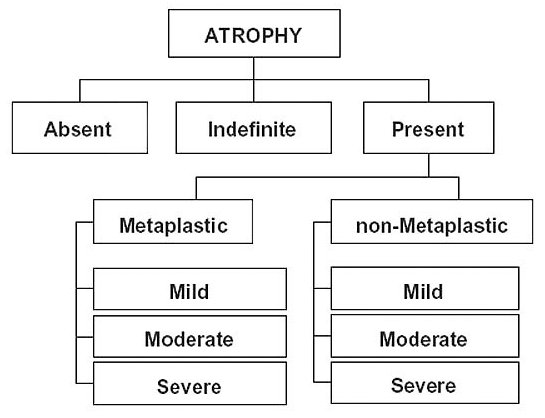 Clasificación de la atrofia gástrica propuesta por el denominado Club de la Atrofia (Rugge y col, 2002). Las categorías de atrofia metaplásica y no metaplásica coexisten habitualmente en el mismo paciente (esquema reproducido con permiso del Prof. RM Genta). ----------------------------------------------------------------------------------------------
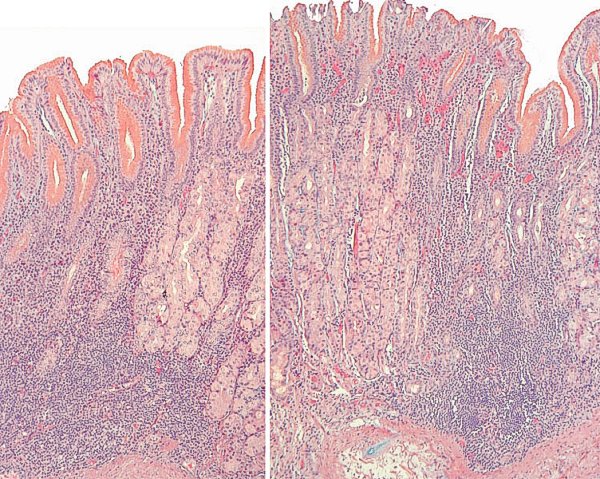 Mucosa de cuerpo gástrico con categoría indefinida para atrofia gástrica. Las glándulas oxínticas quedan separadas y desplazadas por el infiltrado inflamatorio (imagen reproducida con permiso del Prof. RM Genta). ----------------------------------------------------------------------------------------------
La pérdida glandular es seguida de
una sutil cicatriz en la lámina propia, con reemplazamiento fibroso
por haces de colágena y colapso reticulínico del tejido conectivo
interglandular. Estas pequeñas áreas circunscritas de fibrosis residual
de la lámina propia son marcadores de atrofia y pueden pasar
inadvertidas en la tinción de HE, pero son detectables con la tinción
de Gomori.
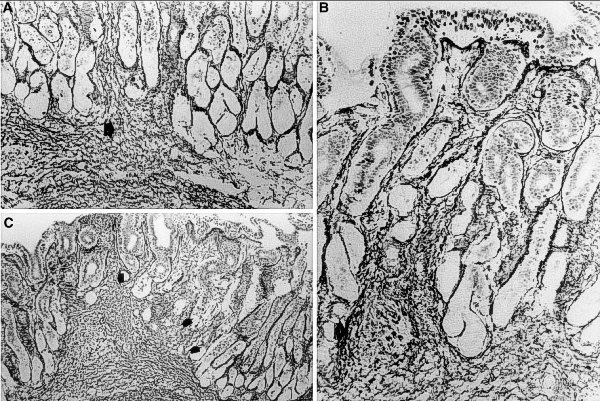 A y B = mucosa antral con pérdida glandular y reemplazamiento fibroso (flecha). C área de pérdida glandular con fibras de reticulina engrosadas y compactadas extendiéndose en el tejido interglandular (flechas). Imágenes cedidas por el Prof. Ricardo Drut y reproducidas con su permiso. ----------------------------------------------------------------------------------------------
La categoría de atrofia asociada con
cambio metaplásico es fácilmente reconocible. Cuando existe solo
atrofia sin MI el grado 1 (atrofia leve), puede pasar inadvertido. La
mucosa antral atrófica presenta menor grado de ramificación glandular y
puede observarse una expansión del tejido conectivo interglandular.
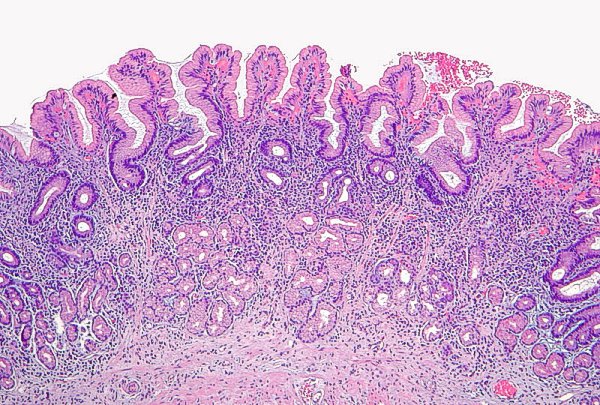 Atrofia no metaplásica de grado leve de mucosa antral (imagen reproducida con permiso del Prof. RM Genta). ----------------------------------------------------------------------------------------------
En la mucosa de cuerpo gástrico la atrofia se expresa en un acortamiento de los túbulos oxínticos y un ensanchamiento del espacio interglandular. En el grado 1 (atrofia leve), puede puede observarse sólo una pequeña pérdida de glándulas oxínticas reemplazada por tejido fibroso. 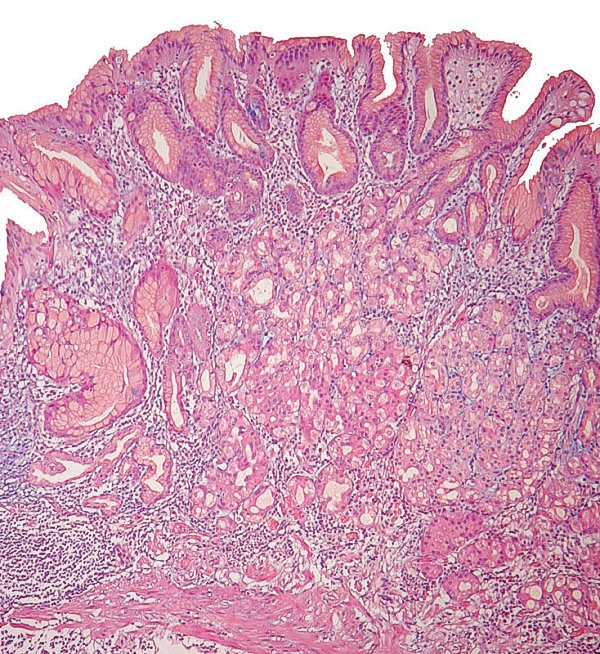 Atrofia no metaplásica de grado leve en mucosa de cuerpo gástrico. Se detecta dilatación foveolar y leve hiperplasia epitelial en la invaginación del epitelio foveolar del ángulo izquiedo de la imagen (reproducida con permiso del Prof. RM Genta). ----------------------------------------------------------------------------------------------
 Atrofia no metaplásica de grado moderado en mucosa de cuerpo gástrico (imagen reproducida con permiso del Prof. RM Genta). ----------------------------------------------------------------------------------------------
En algunos casos de atrofia severa,
la mucosa de cuerpo gástrico puede perder toda semejanza con su
arquitectura original y adoptar una apariencia idéntica a la de la
mucosa antral normal. En estos casos el diagnóstico de atrofia sólo es
posible con una precisa información topográfica del endoscopista. Esta
es la razón por la que las 5 muestras procedentes de las 3
localizaciones indicadas en el Sistema Sydney revisado (Dixon y
col, 1996), deben ser siempre remitidas en 3 envases independizados y
correctamente etiquetados (¡¡ nunca con la etiqueta en la tapa !!).
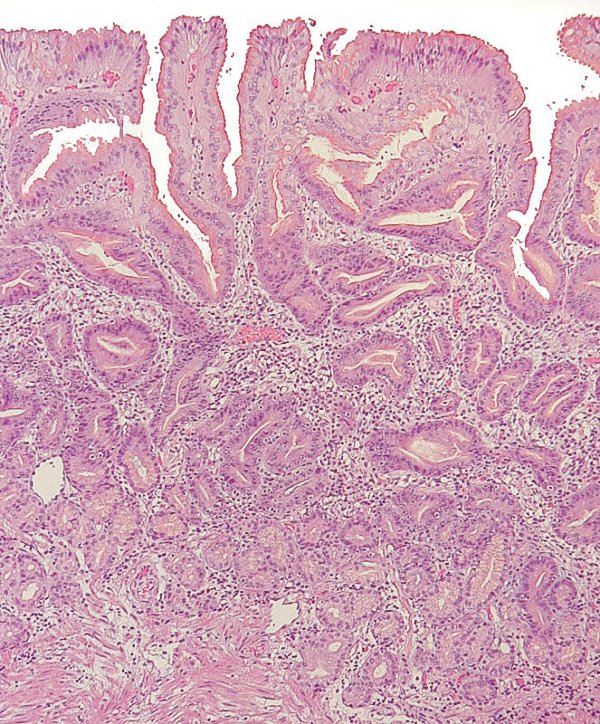 Atrofia no metaplásica de grado severo en mucosa de cuerpo gástrico, con hiperplasia leve del epitelio de los cuellos glandulares (imagen reproducida con permiso del Prof. RM Genta). ----------------------------------------------------------------------------------------------
En un reciente y acertado artículo
(Rugge y Genta, 2005), se propone realizar una gradación y
estadiaje de las gastritis crónicas, de forma análoga al que
habitualmente se efectúa con las biopsias hepáticas. Los propios
investigadores que trabajan en patología gástrica, no encuentran
adecuado aplicar los criterios del Sistema Sydney en la práctica
asistencial por el elevado tiempo que consume respecto de la relevancia
clínica (Owen, 2003). La propuesta de gradación y estadiaje
(Rugge y Genta, 2005), simplifica e integra los parámetros del
Sistema Sydney, al no tener en cuenta el tipo de inflamación y quedar
englobada la MI en la atrofia.
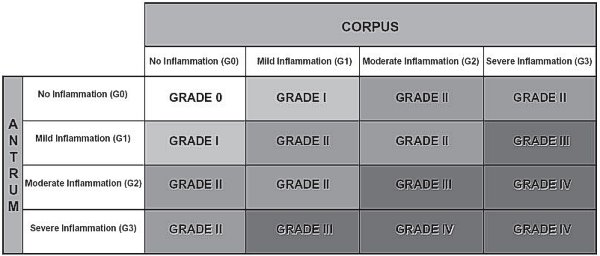 Gradación de la atrofia gástrica recientemente propuesta (Rugge y Genta, 2005). Esquema reproducido con permiso del Prof. RM Genta. ----------------------------------------------------------------------------------------------
En la gradación (0-IV) se combina la intensidad del infiltrado inflamatorio de ambas localizaciones antral y de cuerpo (G0-G3), correspondiendo el grado V a G3+G3, pero se realiza de forma equilibrada y simétrica. Es decir el grado III corresponde tanto a G3 antro + G1 cuerpo, como a G1 antro + G3 cuerpo, por lo que no se tiene en cuenta en el valor final de la gradación el mayor riesgo de carcinogénesis atribuible a la inflamación de predominio en cuerpo (Owen, 2003). En cuanto al estadiaje (0-IV), combina el grado de atrofia de la mucosa (metaplásica y no metaplásica), de ambas localizaciones antral (incluyendo en antro la incisura angular), y de cuerpo (G0-G3). 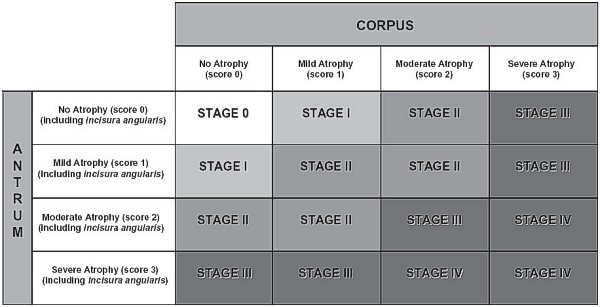 Estadiaje recientemente propuesto de la atrofia gástrica (Rugge y Genta, 2005). Esquema reproducido con permiso del Prof. RM Genta. ----------------------------------------------------------------------------------------------
|
|
|
|
La proliferación celular en los
cuellos glandulares es muy elevada ya que debe atender a la demanda de
renovación de la célula foveolar, del epitelio glandular mucoso antral
y del epitelio glandular especializado. Las células parietales y
principales del epitelio especializado, precisan de una maduración más
prolongada y compleja, por lo que en ocasiones su pérdida no llega a
ser totalmente sustituida. La sustitución del epitelio lesionado
(células parietales y principales), puede ser a expensas de (1) células
mucosas de tipo antral (metaplasia antral o antropilórica), (2)
enterocitos, células caliciformes y de Paneth (MI completa), o (3)
finalmente pueden participar células con características morfológicas y
funcionales ambiguas entre la célula absortiva intestinal de borde en
chapa y la caliciforme (son las células indeterminadas o inmaduras que
morfológicamente definen la MI incompleta). No existe acuerdo si los
distintos tipos de MI son entidades independientes (Filipe y Jass,
1986), o constituyen un espectro con gradiente dependiente de la
extensión de la MI (Stemmermann, 1994), si bien esta última
hipótesis parece la más congruente.
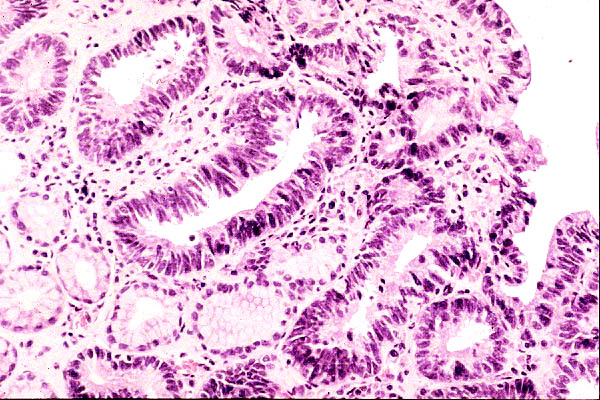
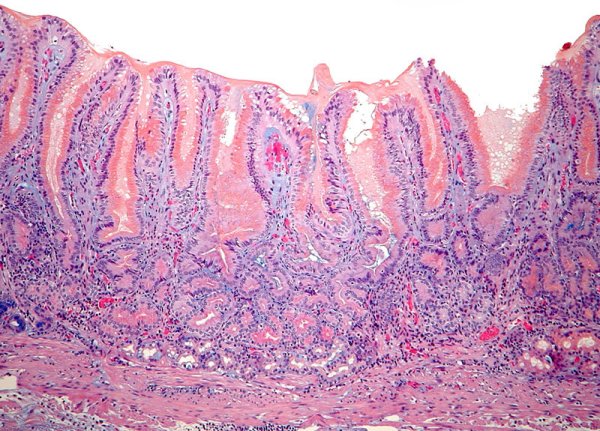 Mucosa de cuerpo gástrico con metaplasia de tipo antral , antropilórica o pseudopilórica. Glándulas mucosas de tipo antral han reemplazado a glándulas oxínticas (imagen reproducida con permiso del Prof. RM Genta). ----------------------------------------------------------------------------------------------
Existen numerosas clasificaciones de
la MI. Hay autores que distinguen hasta 2 y 3 tipos de MI completa en
función de la proporción de células caliciformes y de enterocitos
(Segura y Montero, 1983), así como de la composición de mucinas
(Barachini y col, 1991). La MI completa es conocida también como MI
tipo I (Jass y Filipe, 1979). Dentro de las MI incompletas se han
distinguido los tipos II y III en dependencia de la menor o mayor
proporción de celularidad inmadura, de la menor o mayor distorsión
arquitectural glandular y de la ausencia o presencia de secreción de
sulfomucinas por parte de la celularidad columnar inmadura,
respectivamente (Filipe y Jass, 1986).
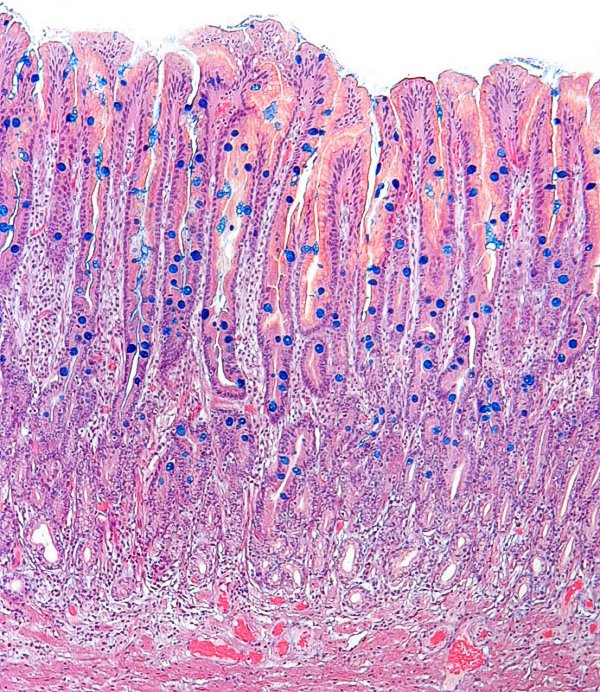 Mucosa de cuerpo gástrico con atrofia severa y MI de tipo completo (imagen reproducida con permiso del Prof. RM Genta). ----------------------------------------------------------------------------------------------
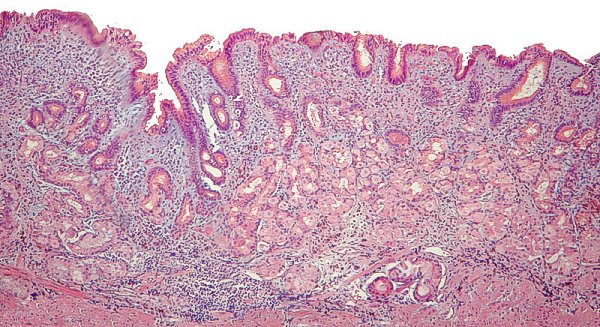
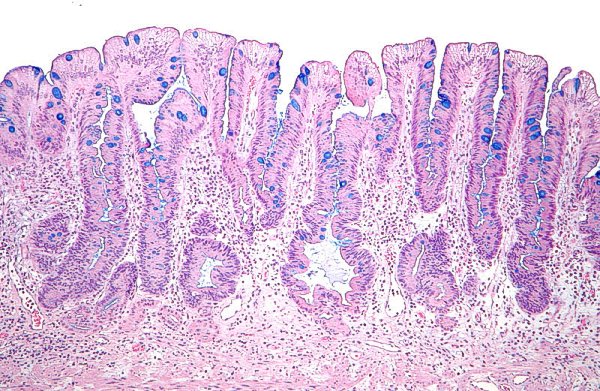 Mucosa gástrica antral con atrofia severa y MI incompleta (imagen reproducida con permiso del Prof. RM Genta). ----------------------------------------------------------------------------------------------
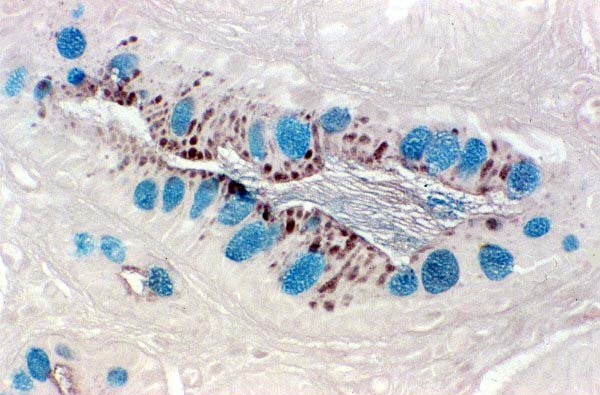 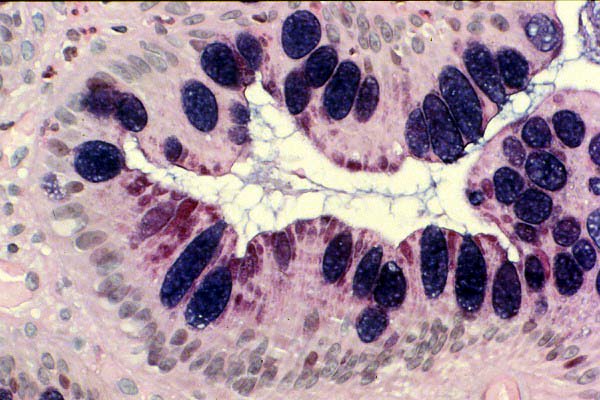  Tinción de AA-PAS a pH 2.5. --------------------------------------------------------------------------------------------
La tinción de HID-AA que discrimina
dentro de las mucinas ácidas las sulfomucinas del resto de mucinas
ácidas prácticamente no se utiliza, ya que a pesar de que la MI tipo
III es la que mayor riesgo precursor de CG presenta (Filipe y Jass
1986), el perfil histoquímico de mucinas ha sido superado por la
inmunohistoquímica, como veremos más adelante (Jass, 2000). No
obstante la tinción de AA-PAS a pH 2.5 sigue siendo de gran ayuda, ya
que detecta las MI incompletas cuyas células columnares inmaduras
presentan un fenotipo mixto con mucinas neutras y ácidas, el cual se
aproxima al imunofenotipo mixto (gástrico e intestinal) definido
mediante antígenos MUC. (Jass, 2000). Inicialmente se aceptó que la
MI tipo II sería un paso intermedio en la evolución de la MI tipo I a
la MI tipo III(Filipe y Jass 1986), si bien posteriormente se ha
cuestionado esta transición.
|
|
|
|
Entre los primeros investigadores que
centraron su atención en la DG figura Takeo Nagayo, quien a mediados
del pasado siglo empezó a estudiar la mucosa que rodeaba los CG,
clasificando en 1971 las lesiones en 5 grados: ausencia de atipia,
atipia leve, atipia límite, probable cáncer y CG (Nagayo, 1986).
1. - ALTERACIONES CITOLOGICAS:
Anomalías de forma, tamaño, configuración y orientación, tanto en el
núcleo como en el citoplasma. Esta atipia citológica es particularmente
marcada en la DG de tipo metaplásico. En el caso de la DG no
metaplásica, las células permanecerían cuboidales, sin llegar a
transformarse en células intestinales. El tamaño nuclear parece ser el
parámetro que mejor diferencia entre lesión displásica y reactiva.
Además del incremento de la relación núcleo-citoplasma, existe
hipercromasia, pleomorfismo nuclear, pérdida de la polaridad del núcleo
y pseudoestratificación (imagen global que resulta al observar los
núcleos de las células a distinta altura, en lugar de permanecer
uniformemente ordenados en la zona basal).
2. - ANOMALIAS EN LA DIFERENCIACION:
En el epitelio displásico de tipo metaplásico, se evidencia deplección
y desigual distribución de las células caliciformes, desaparición de
las células de Paneth y estratificación de las células epiteliales por
disminución del citoplasma con pobre desarrollo del borde en cepillo.
En el epitelio gástrico no metaplásico, se detecta disminución de la
actividad mucosecretora.
3. - DISTORSION DE LA ARQUITECTURA
GLANDULAR: Agrupaciones de glándulas alargadas con criptas irregulares,
que pueden presentar luces glandulares "espalda contra espalda" en un
patrón cribiforme por proliferación excesiva del epitelio y
desaparición del estroma.
En función de la intensidad de estos
3 grupos de parámetros, la DG se clasificó en leve, moderada y severa.
No obstante, en 1984, el Grupo Internacional de Estudio del CG (ISGGC)
propuso clasificar las lesiones en solamente 2 niveles: DG de bajo
grado y de alto grado. La simplificación facilitó la reproducibilidad
de la clasificación, pero el diagnóstico de DG continuó siendo
subjetivo (Misdraji y Lauwers, 2002).
 DG de alto grado que alcanza el epitelio foveolar superficial ------------------------------------------------------------------------------------------------- La DG de alto grado es la menos frecuente y parece representar un fenómeno muy tardío en el proceso de carcinogénesis gástrica (Sipponen, 1990). La DG sobre mucosa con MI es más frecuente que la DG no metaplásica, cuya principal característica es el reemplazamiento de la células diferenciadas que revisten las glándulas por otras células indiferenciadas con variables grados de atipia citológica, pero sin distorsión arquitectural. La severidad de la DG no metaplásica viene dada por (1) la mayor o menor extensión de la afectación desde la zona proliferativa de los cuellos glandulares hasta la porción profunda de las criptas, y (2) por el grado de pleomorfismo celular (Ghandur-Mnaymneh y col, 1988). En 1979 Cuello introduce criterios cualitativos y establece 2 niveles para cada uno de los 2 grandes grupos de DG: DG hiperplásica de bajo y alto grado, DG adenomatosa de bajo y alto grado (Cuello y col, 1979). Es la primera vez que que se introducen criterios, no solamente cuantitativos sino también cualitativos, en la clasificación de la DG. La propuesta de Cuello es recogida por Jass, quien a la DG de patrón adenomatoso denomina tipo I y a la de patrón hiperplásico tipo II (Jass 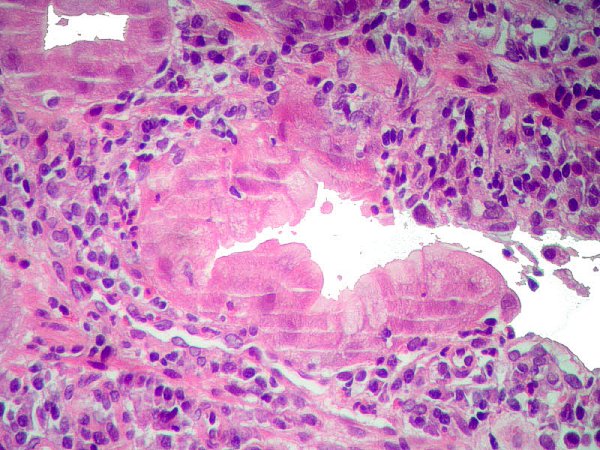 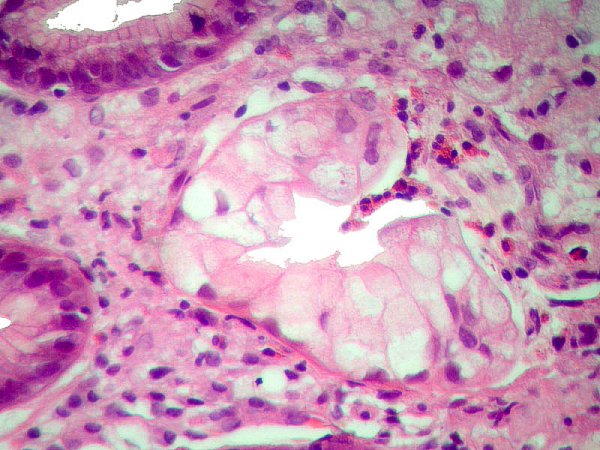 Imágenes interpretadas como cambio proliferativo hiperplásico de patrón globoide, no definitivo de DG (Fatima Carneiro). ---------------------------------------------------------------------------------------------- 1.- negativo para DG (se incluyeron aquí las MI incompletas). 4.1.- DG de alto grado. 4.2.- carcinoma "in situ". 4.3.- s 5.1.- carcinoma intramucoso. 5.2.- carcinoma submucoso o invasor más profundo. 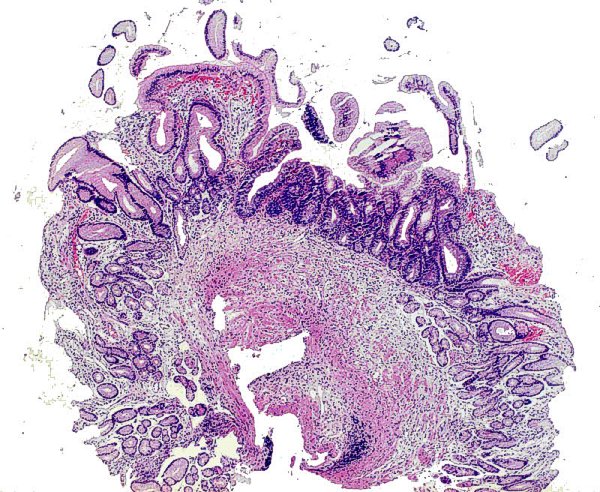 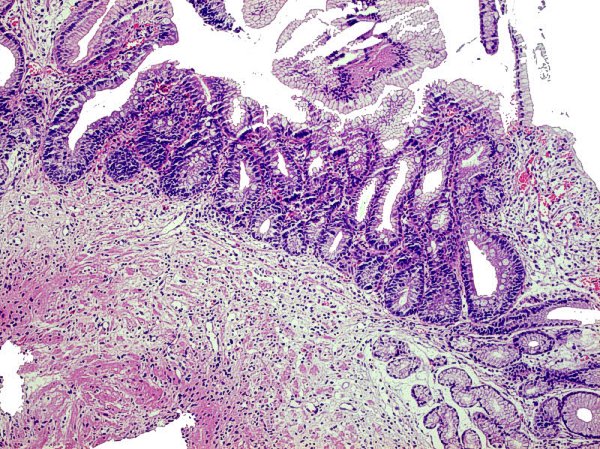 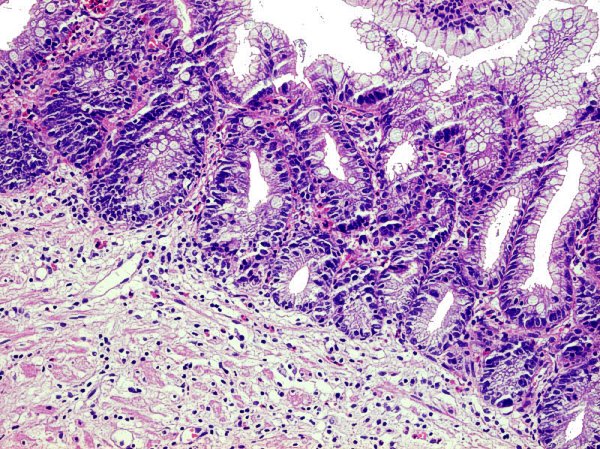 
|
|
|
|
PATOLOGÍA MOLECULARPOLIMORFISMOS Y MUTACIONESLa base molecular de la carcinogénesis por etapas (proceso de LPCG), radica en el daño genético no letal que sufre el epitelio. Este daño genético (mutación), suele ser adquirido (línea celular somática), e inducido por agentes ambientales (Hp). Los polimorfismos son variantes alélicas en línea germinal. La interacción de agentes ambientales con los factores del huésped (polimorfismos responsables de la predisposición genética), va a modular la evolución de la LPCG. HELICOBACTER Y MARCADORES DE SUSCEPTIBILIDAD GENÉTICALa progresión de la LPCG es variable en cada indivíduo y depende de una compleja interacción entre factores ambientales y de susceptibilidad genética. La susceptibilidad genética para el CG viene determinada por variaciones alélicas o polimorfismos de los genes que regulan, entre otros, los siguientes aspectos:1.- la respuesta inflamatoria frente a la infección por Hp. 2.- los mecanismos de protección de la mucosa gástrica frente al Hp y otros agentes carcinógenos. 3.- la capacidad detoxificante de agentes carcinógenos. 4.- el equilibrio entre daño oxidativo y la protección frente al mismo. 5.- los mecanismos de reparación del DNA. 6.- los procesos de proliferación, maduración y adhesión celular. Desarrollar cada uno de estos puntos escapa a las pretensiones de esta revisión. Solamente se comentarán algunos aspectos relacionados con la respuesta inflamatoria y la infección por Hp. La modulación genética de la síntesis de prostaglandinas (PTGGS1 y PTGGS2), interviene en la progresión de la LPCG y podría explicar el efecto protector frente al CG tanto del consumo de aspirina (Farrow y col 1998), como de borraja (González y col, 1993). La gastritis por Hp evoluciona hacia la cronicidad en 2 rutas distintas y divergentes. Unos pacientes desarrollan atrofia gástrica y finalmente úlcera gástrica y/o CG. Otros pacientes no experimentan atrofia gástrica y finalmente desarrollan úlcera duodenal o linfoma MALT (Owen, 2003). El Hp coloniza la mucosa gástrica de casi la mitad de la población mundial. La infección, por lo general adquirida en la infancia, dejada evolucionar libremente puede persistir durante toda la vida del huésped. Aunque el riesgo varia con la edad, área geográfica y etnia, entre el 15% y el 20% de las personas infectadas puede seguir la ruta de la úlcera péptica y menos del 1% llegarán a desarrollar CG. Afortunadamente la mayor parte de los pacientes infectados con Hp no desarrollará ninguna de estas patologías (Stoicov y col, 2004). Uno de los argumentos más esgrimidos por los detractores de la infección por Hp como causa de CG, es su poca frecuencia entre la población africana, cuando Africa es el continente con mayores tasas de infección por Hp. Es el denominado enigma africano y parece ser debido al impacto que otros agentes infecciosos y parasitarios, pueden tener en la respuesta inflamatoria del huésped. Determinadas infestaciones helmínticas (Heligmosomoides polygyrus), inducen un patrón de respuesta inflamatoria con citoquinas protectoras de la atrofia (Fox y col, 2000). Las frecuentes infecciones e infestaciones en la población africana, parecen alterar la respuesta inflamatoria del huésped hacia el Hp (Stoicov y col, 2004). Las cepas de Hp con mayor grado de virulencia y que provocan mayor daño tisular son las capaces de sintetizar las citotoxinas Cag A, citotoxina vacuolizante Vac A y el factor activador de los neutrófilos NAF. Tanto los pacientes que siguen la ruta del CG como los que siguen la ruta de la úlcera péptica, son infectados con cepas virulentas en mayor proporción que los pacientes que no desarrollan patología (Atherton, 1998). Hasta la fecha no se ha detectado ningún factor en el Hp selectivamente asociado con el CG y que no siga la ruta de la úlcera dudodenal. Recientemente, entre 14 genes que codifican la virulencia del Hp, se ha detectado el denominado gen promotor de la úlcera duodenal (dupA), selectivamente asociado con la ruta de evolución a úlcus péptico y no hacia CG (Lu y col, 2005). Determinadas variantes alélicas de los genes LTA y TNF así como algunos polimorfismos del gen de la interleukina 1-beta, (IL)-1ß, podrían ser la causa de que unas personas infectadas por Hp desarrollen úlcera péptica y otras CG (González y col, 2002). Distintos polimorfismos funcionantes en los genes IL-1ß, IL-1RN, IL-10 y TNF-A se han encontrado asociados con el riesgo de CG, al menos en poblaciones americana, escocesa, portuguesa y china (Yang y col, 2004). Por el contrario no se han detectado estos polimorfismos en pacientes con úlcera duodenal (García-González y col, 2005). La interleukina (IL)-1ß presenta el efecto inhibidor de la secreción ácida gástrica de mayor magnitud conocida. Se estima que es 100 veces más potente que los fármacos inhibidores de la bomba de protones y unas 6000 veces más potente que los antagonistas H2. Los polimorfismos genéticos que incrementan la producción de IL-1ß, responsable de la hipoclorhidria y de una respuesta inflamatoria predominante en cuerpo gástrico, conducirían a un mayor riesgo de CG no cardial. Los pacientes que no presentan los referidos polimorfismos, no ven afectada su secreción ácida gástrica o incluso está incrementada tras la infección por Hp. En ellos, la gastritis es de predominio antral y se asocia con úlcera péptica (El-Omar y col, 2001). Como se comentó en el capítulo de inflamación, el patrón topográfico de las gastritis (intensidad de la inflamación en mucosa de cuerpo, respecto a la de antro), pudiera ser un factor determinante para establecer el riesgo de carcinogénesis (Owen, 2003). MUTACIONES DEL GEN CDH1 Y SU EXPRESIÓN FENOTÍPICAEl gen CDH1 regula la síntesis y expresión de la E-cadherina. La E-cadherina forma parte de una familia de proteínas de adhesión celular y su expresión resulta clave en los procesos de diferenciación y migración celular durante el desarrollo embrionario (Guilford, 1999). Las mutaciones en línea germinal del gen CDH1 son las responsables de aproximadamente el 30% de los CG hereditarios de patrón difuso (HDGC = Síndrome Hereditario del Carcinoma Gástrico Difuso). La primera familia estudiada en la que se detectó mutación del gen CDH1 era de origen Maorí y presentaba una penetrancia del 70% (Guilford y col, 1998). Tras estudiar 32 familias portuguesas con historia familiar de CG se detectó 1 familia portadora de la mutación del gen CDH1 que cumplía todos los criterios de HDGC y otra familia con criterios incompletos de HDGC era portadora de una mutación del gen p53 en línea germinal, por lo que los autores del estudio recomiendan explorar la mutación del gen p53 en sujetos con sospecha de HDGC que no presenten mutación del gen HDCG (Oliveira y col, 2004). Estas mutaciones son raras, se han descrito en familias de Nueva Zelanda y Portugal. La probabilidad del desarrollo de CG difuso en miembros jóvenes de estas familias portadores de mutaciones heterozigotas del gen CDH1 es muy elevada, ya que presentan un patrón autosómico dominante de herencia con una penetrancia del 80% (Huntsman y col, 2001). Por esta razón se ha preconizado la gastrectomía total profiláctica, habiéndose encontrado en todos los casos intervenidos focos de CG difuso, tras incluir entre 150 y 250 bloques de parafina (Lewis y col, 2001). El seguimiento estricto mediante cromoendoscopia y biopsias múltiples de las áreas sospechosas marcadas, podría representar una alternativa frente a la gastrectomía. La cromoendoscopia es una técnica poco extendida, precisa entrenamiento por parte del endoscopista, y existe cierta polémica sobre su rendimiento. Además de las mutaciones del gen promotor de la E-cadherina (CDH1), recientemente se han descrito determinados polimorfismos de este gen asociados con una mayor frecuencia de la infección por Hp (Liu y col, 2005). Estos pacientes requerirían la infección por Hp para la inactivación del gen CDH1 y dado que los polimorfismos son también en línea germinal, explicaría el papel del Hp en los estadíos iniciales de la LPCG. La pérdida de expresión de E-cadherina durante la carcinogénesis gástrica condicionaría que unos tumores fueran de patrón intestinal (expresión parcialmente conservada) y otros de patrón difuso (pédida de expresión). Adicionalmente, algunos investigadores han llamado la atención del papel de la expresión de la E-cadherina en las áreas de metaplasia intestinal, ya que el fenómeno podría representar un válido marcador intermedio de carcinogénesis gástrica (Zullo y col, 2004). Lamentablemente también la mucosa de intestino delgado normal puede presentar pérdida de expresión de E-cadherina y los patrones de normalidad en la expresión de E-cadherina en la mucosa gástrica precisarían estar mejor definidos. CD44 es otra molécula de adhesión celular candidata a jugar un papel en la evolución de la LPCG (Tahara, 2004).
PROTOONCOGENES Y GENES ONCOSUPRESORES
La proliferación celular está
regulada por 2 tipos de genes. Los protooncogenes activan el
crecimiento y los genes oncosupresores lo inhiben. Se denominan
proteínas salvajes las codificadas por estos genes normales. La
mutación de ambos tipos de genes dan lugar a oncogenes y anti-oncogenes
(genes oncosupresores). Las proteínas codificadas por ellos se
denominan proteínas mutantes. Todo proceso de carcinogénesis implica
una secuencia de múltiples mutaciones acumulativas, tanto en
protooncogenes como en genes oncosupresores.
Una mutación activadora en tan sólo uno de los alelos de un protooncogén, lo convierte en un oncogén capaz de inducir la transformación neoplásica de la célula. Se trata de una mutación dominante con ganancia de función. Por el contrario, para que la mutación de un gen oncosupresor implique pérdida de función, ha de afectar a los 2 alelos. Son mutaciones recesivas y cuando afectan a sólo uno de los alelos debe producirse una pérdida ulterior de la heterozigosidad (LOH), para que se produzca el efecto oncogénico. El CG presenta a menudo pérdida o inactivación de los genes oncosupresores p53, APC y DCC. La frecuencia de estas alteraciones difieren considerablemente según se trate de CG de patrón intestinal o de patrón difuso. En el 60% de los CG se han detectado mutaciones del gen p53 y LOH en el cromosoma 17p (locus p53). Existe todo un espectro de mutaciones p53 y las mutaciones de este gen en los CG de patrón intestinal difieren de las observadas en los CG de patrón difuso. Algunos estudios han sugerido que las mutaciones p53 se sucederían en estadíos avanzados de LPCG, detectándose preferentemente en DG de alto grado y no en DG de bajo grado (Tahara, 1993). Otros autores han encontrado mutaciones p53 en lesiones de MI (Mourad y col, 2004). En modelos experimentales con ratones transgénicos portadores de mutaciones APC y p53 se ha seguido la evolución de MI a CG (Mutoh y col, 2004), evidenciando el papel precursor de CG intrínseco a la lesión metaplásica que había sido cuestionado por algunos autores (El-Zimaity y col, 2001) En el 60% de los CG precoces de patrón intestinal se detecta LOH en el cromosoma 5q (gen APC), siendo mucho más baja la frecuencia de esta alteración en el CG difuso. En la mitad de los CG de patrón intestinal se ha encontrado LOH del cromosoma 18q (locus DCC), alteración no detectada en los CG difusos. Otras alteraciones selectivamente asociadas al CG de patrón intestinal son: mutaciones K-ras (aunque su incidencia es baja si se compara con los carcinomas de colon páncreas), amplificación de c-erbB2 y delección de Bcl-2 (Tahara, 1993). Bcl-2 es un gen antiapoptótico detectado en lesiones de MI, también candidato a modular la LPCG (Topal y col, 2004). Las mutaciones K-ras, aunque poco frecuentes tanto en CG como en MI (10%), parecen acontecer en las primeras fases de la LPCG y s presencia podría predecir la evolución hacía CG (Hao y col, 1998). Tanto la expresión de c-Myc como de telomerasa, están elevadas en el CG y el las LPCG. La magnitud de esta expresión parece presentar un gradiente creciente con la progresión de la LPCG, que estaría influenciado por la infección de Hp (Zhang y col, 2004). TELOMERASACuando una célula se divide y duplica su material genético, sus telómeros (extremos de cromosomas constituidos por DNA repetitivo), pueden acortarse o crecer según las circunstancias. El acortamiento camina hacia el envejecimiento y el alargamiento hacia la inmortalidad. La actividad telomerasa mantiene la longitud del telómero evitando su acortamiento. Células que acumulan demasiadas mutaciones suelen tener telómeros muy cortos y la telomerasa está inactiva. La reactivación de la actividad telomerasa proporciona inmortalidad a la célula neoplásica.Ya en su revisión anterior de 1993, Tahara hacía referencia a las anomalías en la reducción del telómero detectadas en la MI (Tahara, 1993). La actividad intensa de la telomerasa se asocia con la expresión de hTERT (human telomerase reverse transcriptase), presente en la mayor parte de los CG y también en la MI. La infección por Hp parece actuar como un potente desencadenante de la proliferación de células hTERT positivas precursoras de MI (Tahara, 2004). Probablemente la reactivación de la actividad telomerasa es un evento precoz de la transformación neoplásica en los CG tanto proximales como distales (Gulmann y col, 2005). MARCADORES DE DIFERENCIACIÓN
Algunos investigadores han señalado
que durante la evolución de la DG hacia CG (Testino y col, 2002),
parece existir una pérdida progresiva de marcadores de diferenciación
gástrica (pepsinógeno C y antígeno M1 gástrico foveolar), así como una
progresiva expresión de marcadores de diferenciación enteropancreática
(antígenos intestinal CAR-5 y pancreático DU-PAN-2), con relativa
independencia de la morfología lesional (DG de alto o bajo grado).
Los patrones de expresión de varios genes que regulan la síntesis de mucinas, proporcionan actualmente uno de los mejores instrumentos para evaluar los trastornos de diferenciación durante la evolución de la LPCG (Jass, 2000). Las mucinas son glucoproteínas de elevado peso molecular en las que pueden distinguirse 3 dominios o porciones con capacidad antigénica: La región central o "core" de naturaleza peptídica, la región intermedia formada por unidades de disacáridos y la región periférica caracterizada por los antígenos de grupo sanguíneo A, B, H y Lewis Le(a) / Le(b) (Feizi y col, 1984). La clasificación de la MI se realizó según patrones histoquímicos (Filipe y Jass, 1986), centrados en el pH (mucinas ácidas y neutras), y en la presencia entre las ácidas de grupos sulfato (sulfomucinas) y de acetilación siálica (sialomucinas). Paralelamente se empezaron a ensayar antígenos de mucinas. El LIMA (large instestine mucin antigen) se encontró asociado con la progresión de la LPCG y se propuso utilizarlo para distinguir la DG (LIMA+), de los cambios reactivos en la MI (Filipe y col, 1988). En palabras textuales de JR Jass, impulsor junto con Isabel Filipe de la histoquímica de mucinas (Filipe y Jass, 1986), "el clonado y secuenciación de los genes de mucinas epiteliales ha convertido el arte visualmente atractivo de la histoquímica de mucinas, en un instrumento de precisión científica para la exploración de los mecanismos biológicos que subyacen a las alteraciones de crecimiento y diferenciación (Jass, 2000). Entre los genes que codifican las porciones centrales "core" de las mucinas gastrointestinales, los más representativos para el estudio de alteraciones en los procesos de maduración son:
La
mucosa gástrica normal expresa MUC1 y MUC5AC en el epitelio foveolar y
MUC6 en las glándulas. Estos 3 antígenos de mucinas definen el fenotipo
gástrico. MUC2 define el fenotipo intestinal (Jass, 2000). La MI
completa presenta un fenotipo intestinal puro, sin evidencia de
expresión de marcadores gástricos (MUC1, MUC5AC y MUC6). La MI
incompleta co-expresa ambos grupos de marcadores en variable
proporción, por lo que presenta un fenotipo mixto: gástrico e
intestinal. En la práctica asistencial, la tinción de AA-PAS a pH 2,5
recomendada por el Sistema Sydney revisado (Dixon y col, 1996),
continúa siendo una tinción histoquímica válida para la detección del
fenotipo mixto de mucinas gástricas e intestinales, en la celularidad
inmadura de la MI incompleta. Los distintos polimorfismos del gen MUC1 parecen presentar variable grado de susceptibilidad genética. Los polimorfismos homozigotos para alelos pequeños del gen MUC1 podrían estár asociados con un mayor riesgo de CG. Estos polimorfismos se detectan en algunas gastritis crónicas y preferentemente en gastritis atróficas y lesiones de MI incompleta. En las MI completas predominan los polimorfismos de homozigotos de alelos grandes. En lesiones sin atrofia ni MI, los polimorfismos heterozigotos (Silva y col, 2001).
------------------------------------------------------------------------------------------------- ALTERACIÓN EN LA EXPRESIÓN DE ANTÍGENOS LEWIS EN MUCOSA GÁSTRICALos antígenos de grupo sanguíneo Lewis corresponden a glucoproteínas integradas en las secreciones mucosas, que juegan un importante papel en los fenómenos de reconocimiento, diferenciación y cohesión celular. Su síntesis está regulada por 2 genes, el gen Lewis y el gen secretor. Los antígenos Lewis de superficie se expresan sólo en el epitelio foveolar superficial, pero en condiciones normales no en las glándulas profundas. Existen 2 antígenos Lewis de superficie, el Le(a) y Le(b). Los individuos con ambos genes, secretor y Lewis, expresan el antígeno Le(b), mientras que los sujetos que carecen de gen secretor expresan el antígeno Le(a). Los individuos sin ninguno de los 2 genes no expresan ninguno de los 2 antígenos (Torrado y col, 1990).Caben sólo 3 posibilidades o fenotipos Lewis: 1.- fenotipo Lewis a-b+ SON TODOS SECRETORES (70% de la población).
2.- fenotipo Lewis a+b- SON TODOS NO SECRETORES (20% de la población).
3.- fenotipo Lewis a-b- HAY SECRETORES Y NO SECRETORES (10% de la población).
No existe el fenotipo Lewis a+b+.
El fenotipo Lewis se determina mediante anticuerpos frente a ambos antígenos y sólo es preciso una mínima porción de epitelio foveolar gástrico superficial. El estado secretor se determina mediante los grupos antigénicos ABH. Existen 3 anticuerpos, anti A, anti B y anti H. La presencia de antígenos ABH en el epitelio foveolar define el fenotipo secretor. No obstante, como se desprende de las 3 posibilidades de fenotipo Lewis, ambos genes están muy relacionados y sólo en el fenotipo Lewis a-b- sería indispensable recurrir al grupo ABH para conocer el estado secretor, por lo que solamente con los anticuerpos anti Le(a) y anti Le (b) puede fenotiparse con garantías el 90% de la población. El anticuerpo frente a Lewis b (anti Le (b)), se fija sobre el epitelio foveolar y un mínimo fragmento es suficiente para conocer si es positivo y por tanto susceptible de estudio. Solamente en los pacientes con fenotipo Lewis a-b+ PUEDE ESTUDIARSE SI EXISTE O NO UNA EXPRESIÓN ANÓMALA de Le(a). La expresión anómala de Le(a) se puede manifestar:
1.- sólo las células caliciformes (ALTERACION TIPO I)
2.- caliciformes y debilmente columnares (ALTERACION TIPO II)
3.- fuerte positividad en ambas, caliciformes y columnares (ALTERACION TIPO III)
La célula columnar es patrimonio exclusivo de la MI incompleta, luego las alteraciones II y III se darán sólo en MI incompletas. La MI incompleta con alteraciones en la expresión Lewis tipos II y III, parece presentar mayor riesgo que cuando no hay alteración (Torrado, 1992a). La expresión anómala del antígeno Lewis(a) en pacientes con fenotipo Lewis (a-b+), se ha asociado estrechamente con la severidad de la lesión precursora de CG, especialmente la DG y la MI tipo III, en poblaciones de alto riesgo de CG, tanto en estudios transversales (Torrado y col, 2000), como de seguimiento (Torrado y col, 1992b). Recuérdese que la expresión anómala de Le(a) es sólo estudiable en sujetos Lewis a-b+ (todos ellos secretores). Los no secretores (Lewis a+b- y parte de Lewis a-b-), también parecen presentar mayor riesgo de CG, por lo que podría establecerse el siguiente orden decreciente de riesgo de CG: 1.- Secretores con alteraciones II y III.
2.- No secretores.
3.- Secretores con alteración I o sin alteraciones.
Además de los antígenos Lewis de superficie Le(a) y Le(b), expresados en el epitelio foveolar, existen otros 2 antígenos que se expresan en la glándulas Le(x) y Le(y). Algunos estudios que han relacionado la expresión de antígenos Lewis con los distintos de MI no han encontrado diferencias significativas (Silva y col, 2002). Estos mismos estudios han detectado el fenotipo intestinal (MUC2) en la MI completa y el mixto (gástrico+intestinal) en las MI incompletas (tipos II y III). La secreción de sulfomucinas en la célula columnar inmadura metaplásica discrimina entre la MI tipo II (ausencia), y la tipo III (presencia). Como ambos tipos de MI incompleta presentan fenotipo MUC mixto y la secreción de sulfomucinas está asociada con los antígenos Lewis, los autores del estudio concluyen que la ausencia o presencia de sulfomucinas es poco relevante (Silva y col, 2002).
|
|
|
|
Agradecemos a Robert Genta y Ricardo Drut, la cesión para esta revisión de las imágenes proporcionadas. Agradecemos a Fátima Carneiro su ayuda en la interpretación de los casos iconografiados.
|
|
|
|
Atherton JC . H. pylori virulence factors. Br Med Bull. 1998;54(1):105-20. Baracchini P , Fulcheri E, Lapertosa G. Patterns of intestinal metaplasia in gastric biopsies. A comparison of different histochemical classifications. Histochem J. 1991 Jan;23(1):1-9 Carpentieri DF , Wenner W, Liquornik K, Ruchelli E. Significance of lymphoid follicles and aggregates in gastric mucosa of children. Pediatr Dev Pathol. 2000 Mar-Apr;3(2):177-9. Cohen M , Rua EC, Balcarce N, Drut R. Histologic Findings in ''"Ex- Helicobacter pylori''" Gastric Biopsies of Pediatric Patients. Pediatr Dev Pathol. 2005 Jul 14; 8: 1-7. Correa P , Haenszel W, Cuello C, Tannenbaum S, Archer M. A model for gastric cancer epidemiology. Lancet. 1975 Jul 12;2(7924):58-60. Correa P . Precursors of gastric and esophageal cancer. Cancer. 1982 Dec 1;50(11 Suppl):2554-65. Correa P. Chronic gastritis: a clinico-pathological classification.Am J Gastroenterol. 1988 May;83(5):504-9. Correa P. Is gastric carcinoma an infectious disease? N Engl J Med. 1991 Oct 17;325(16):1170-1. Correa P. Human gastric carcinogenesis: a multistep and multifactorial processFirst American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992 Dec 15;52(24):6735-40. Correa P, Yardley JH. Grading and classification of chronic gastritis: one American response to the Sydney system. Gastroenterology. 1992 Jan;102(1):355-9. Correa P. The biological model of gastric carcinogenesis. IARC Sci Publ. 2004;(157):301-10. Cuello C, Lopez J, Correa P, Murray J, Zarama G, Gordillo G. Histopathology of gastric dysplasias: correlations with gastric juice chemistry. Am J Surg Pathol. 1979 Dec;3(6):491-500. Dixon MF, Genta RM, Yardley JH, Correa P. Classification and grading of gastritis. The updated Sydney System. International Workshop on the Histopathology of Gastritis, Houston 1994. Am J Surg Pathol. 1996 Oct;20(10):1161-81. El-Omar EM, Chow WH, Rabkin CS. Gastric cancer and H. pylori: Host genetics open the way. Gastroenterology. 2001 Oct;121(4):1002-4. El-Zimaity HM, Graham DY. Evaluation of gastric mucosal biopsy site and number for identification of Helicobacter pylori or intestinal metaplasia: role of the Sydney System. Hum Pathol. 1999 Jan;30(1):72-77. El-Zimaity HM, Ramchatesingh J, Saeed MA, Graham DY. Gastric intestinal metaplasia: subtypes and natural history. J Clin Pathol. 2001 Sep;54(9):679-83. Feizi T, Gooi HC, Childs RA, Picard JK, Uemura K, Loomes LM, Thorpe SJ, Hounsell EF. Tumour-associated and differentiation antigens on the carbohydrate moieties of mucin-type glycoproteins. Biochem Soc Trans. 1984 Aug;12(4):591-6. Filipe MI, Bogomoletz WV, Dawson P, Fabiani B, Potet F. Intestinal metaplasia subtypes in the assessment of gastric cancer risk. A multicentre prospective study. Gut 1983; 24: 974. Filipe MI, Jass JR. Intestinal metaplasia subtypes and cancer risk. En: "Gastric Carcinoma". Filipe MI, Jass JR, eds. Churchill Livingstone 1986; 87-115. Filipe MI, Barbatis C, Sandey A, Ma J. Expression of intestinal mucin antigens in the gastric epithelium and its relationship with malignancy. Hum Pathol. 1988 Jan;19(1):19-26. Forman D, Kinlen L. Declining incidence of gastric cancer. BMJ. 1991 Jul 27;303(6796):248-9.Fox JG, Beck P, Dangler CA, Whary MT , Wang TC, Shi HN, Nagler-Anderson C. Concurrent enteric helminth infection modulates inflammation and gastric immune responses and reduces helicobacter-induced gastric atrophy. Nat Med. 2000 May;6(5):536-42. Fox JG, Wang TC. Helicobacter pylori--not a good bug after al l! N Engl J Med. 2001 Sep 13;345(11):829-32. Farrow DC, Vaughan TL, Hansten PD, Stanford JL, Risch HA, Gammon MD, Chow WH, Dubrow R, Ahsan H, Mayne ST, Schoenberg JB, West AB, Rotterdam H, Fraumeni JF, Blot WJ. Use of aspirin and other nonsteroidal anti-inflammatory drugs and risk of esophageal and gastric cancer. Cancer Epidemiol Biomarkers Prev. 1998 Feb;7(2):97-102. Garcia-Gonzalez MA, Savelkoul PH, Benito R, Santolaria S, Crusius JB, Pena AS, Lanas A. No allelic variant associations of the IL-1 and TNF gene polymorphisms in the susceptibility to duodenal ulcer disease. Int J Immunogenet. 2005 Oct;32(5):299-306. Genta RM. Recognizing atrophy: another step toward a classification of gastritis. Am J Surg Pathol. 1996;20 Suppl 1:S23-30. Genta RM . Atrophy and atrophic gastritis: one step beyond the Sydney system. Ital J Gastroenterol Hepatol. 1998 Oct;30 Suppl 3:S273-5. Ghandur-Mnaymneh L, Paz J, Roldan E, Cassady J. Dysplasia of nonmetaplastic gastric mucosa. A proposal for its classification and its possible relationship to diffuse-type gastric carcinoma. Am J Surg Pathol. 1988 Feb;12(2):96-114. Gonzalez CA, Sanz JM, Marcos G, Pita S, Brullet E, Saigi E, Badia A, Agudo A, Riboli E. Borage consumption as a possible gastric cancer protective factor. Cancer Epidemiol Biomarkers Prev. 1993 Mar-Apr;2(2):157-8. Gonzalez CA , Sala N, Capella G. Genetic susceptibility and gastric cancer risk. Int J Cancer. 2002 Jul 20;100(3):249-60. Graham DY , Shiotani A. The time to erradicate gastric cancer is now. Gut. 2005 Jun;54(6):735-8. de Grouchy J . The malignant primate? Ann Genet. 1991;34(3-4):137-42. Guilford P , Hopkins J, Harraway J, McLeod M, McLeod N, Harawira P, Taite H, Scoular R, Miller A, Reeve AE. E-cadherin germline mutations in familial gastric cancer. Nature. 1998 Mar 26;392(6674):402-5. Guilford P . E-cadherin downregulation in cancer: fuel on the fire? Mol Med Today. 1999 Apr;5(4):172-7 Guindi M , Ridell RH. The pathology of epithelial pre-malignancy of the gastrointestinal tract. Best Practice & Research Clinical Gastroenterology Vol. 15, No. 2, pp. 191±210, 2001. Gulmann C , Lantuejoul S, Grace A, Leader M, Patchett S, Kay E. Telomerase activity in proximal and distal gastric neoplastic and preneoplastic lesions using immunohistochemical detection of hTERT. Dig Liver Dis. 2005 Jun;37(6):439-45. Haenszel W , Correa P. Developments in the epidemiology of stomach cancer over the past decade. Cancer Res. 1975 Nov;35(11 Pt. 2):3452-9. Hao Y, Zhang J, Lu Y, Yi C, Qian W, Cui J. The role of ras gene mutation in gastric cancer and precancerous lesions. J Tongji Med Univ. 1998;18(3):141-4. Hitchcock CR , Maclean LD, Sullivan WA. The secretory and clinical aspects of achlorhydria and gastric atrophy as precurors of gastric cancer. J Natl Cancer Inst. 1957 Jun;18(6):795-811. Huntsman DG, Carneiro F, Lewis FR, MacLeod PM, Hayashi A, Monaghan KG, Maung R, Seruca R, Jackson CE, Caldas C. Early gastric cancer in young, asymptomatic carriers of germ-line E-cadherin mutations. N Engl J Med. 2001 Jun 21;344(25):1904-9. Jass JR, Filipe MI. A variant of intestinal metaplasia associated with gastric carcinoma: a histochemical study. Histopathology. 1979 May;3(3):191-199. Jass JR. Role of intestinal metaplasia in the histogenesis of gastric carcinoma. J Clin Pathol 1980; 33: 801-810. Jass JR. A classification of gastric dysplasia. Histopathology 1983; 7: 181-193. Jass JR. Mucin core proteins as differentiation markers in the gastrointestinal tract. Histopathology. 2000 Dec;37(6):561-564. Jiménez-Escrig A . Generalidades de Genética. En Manual de neurogenética. Jiménez-Escrig A. Ed Díaz de Santos. Madrid, 2003. Kapadia CR . Gastric atrophy, metaplasia, and dysplasia: a clinical perspective. J Clin Gastroenterol. 2003 May-Jun;36(5 Suppl):S29-36; discussion S61-2. Lauren P . The two histological main types of gastric carcinoma: Diffuse and So-called Intestinal -type carcinoma. An attempt at histo-clinical classification. Acta Pathol Microbiol Scand. 1965;64:31-49. Lewis FR, Mellinger JD, Hayashi A, Lorelli D, Monaghan KG, Carneiro F, Huntsman DG, Jackson CE, Caldas C. Prophylactic total gastrectomy for familial gastric cancer. Surgery. 2001 Oct;130(4):612-7; discussion 617-9. Lovelock JE . New statements on the Gaia theory. Microbiologia 1995 Sep;11(3):295-304. Liu YC , Shen CY, Wu HS, Chan DC, Chen CJ, Yu JC, Yu CP, Harn HJ, Shyu RY, Shih YL, Hsieh CB, Hsu HM. Helicobacter pylori infection in relation to E-cadherin gene promoter polymorphism and hypermethylation in sporadic gastric carcinomas. World J Gastroenterol. 2005 Sep 7;11(33):5174-9. Lu H , Hsu PI, Graham DY, Yamaoka Y. Duodenal ulcer promoting gene of Helicobacter pylori. Gastroenterology. 2005 Apr;128(4):833-48. Maley CC , Reid BJ. Natural selection in neoplastic progression of Barrett's esophagus. Semin Cancer Biol. 2005 Dec;15(6):474-83. Misdraji J , Lauwers GY. Gastric epithelial dysplasia. Semin Diagn Pathol. 2002 Feb;19(1):20-30. Morson BC, Sobin LH, Grundmann E, Johansen A, Nagayo T, Serck-Hanssen. Precancerous conditions and epithelial dysplasia in the stomach. J Clin Pathol 1980; 33: 711-721. Mourad WA, El Husseiny G, Shoukri M, Rezeig M, Chianzantoniou N, Amin T. Biological markers in Helicobacter pylori-associated gastritis and carcinoma: the value of a scoring system. Ann Saudi Med. 2004 Mar-Apr;24(2):112-8. Mutoh H, Sakurai S, Satoh K, Tamada K, Kita H, Osawa H, Tomiyama T, Sato Y, Yamamoto H, Isoda N, Yoshida T, Ido K, Sugano K. Development of gastric carcinoma from intestinal metaplasia in Cdx2-transgenic mice. Cancer Res. 2004 Nov 1;64(21):7740-7. Nagayo T . Histogenesis and Precursors of Human Gastric Cancer. Research and Practice. Springer-Verlag, Tokio, 1986. Oliveira C, Ferreira P, Nabais S, Campos L, Ferreira A, Cirnes L, Alves CC, Veiga I, Fragoso M, Regateiro F, Dias LM, Moreira H, Suriano G, Machado JC, Lopes C, Castedo S, Carneiro F, Seruca R. E-Cadherin (CDH1) and p53 rather than SMAD4 and Caspase-10 germline mutations contribute to genetic predisposition in Portuguese gastric cancer patients. Eur J Cancer. 2004 Aug;40(12):1897-903. Owen DA. Normal histology of the stomach. Am J Surg Pathol. 1986 Jan;10(1):48-61. Owen DA . Gastritis and carditis. Mod Pathol. 2003 Apr;16(4):325-41. Piazuelo MB, Haque S, Delgado A, Du JX, Rodriguez F, Correa P. Phenotypic differences between esophageal and gastric intestinal metaplasia. Mod Pathol. 2004 Jan;17(1):62-74. Ramírez V. Estudio de Lesiones Precursoras de Cáncer Gástrico en la Provincia de Soria. Tesis Doctoral Universidad de Zaragoza, junio-1994. Rugge M, Correa P, Dixon MF, Fiocca R, Hattori T, Lechago J, Leandro G, Price AB, Sipponen P, Solcia E, Watanabe H, Genta RM. Gastric mucosal atrophy: interobserver consistency using new criteria for classification and grading. Aliment Pharmacol Ther. 2002 Jul;16(7):1249-59. Rugge M, Genta RM. Staging and grading of chronic gastritis. Hum Pathol. 2005 Mar;36(3):228-33. Schlemper RJ, Kato Y, Stolte M. Review of histological classifications of gastrointestinal epithelial neoplasia: differences in diagnosis of early carcinomas between Japanese and Western pathologists. J Gastroenterol. 2001 Jul;36(7):445-56. Segura DI, Montero C. Histochemical characterization of different types of intestinal metaplasia in gastric mucosa. Cancer. 1983 Aug 1;52(3):498-503. Silva F , Carval ho F, Peixoto A, Seixas M, Almeida R, Carneiro F, Mesquita P, Figueiredo C, Nogueira C, Swallow DM, Amorim A, David L. MUC1 gene polymorphism in the gastric carcinogenesis pathway. Eur J Hum Genet. 2001 Jul;9(7):548-52. Silva E , Teixeira A, David L, Carneiro F, Reis CA, Sobrinho-Simoes J, Serpa J, Veerman E, Bolscher J, Sobrinho-Simoes M. Mucins as key molecules for the classification of intestinal metaplasia of the stomach. Virchows Arch. 2002 Mar;440(3):311-7. Epub 2001 Dec 15. Sipponen P . Gastric dysplasia. Curr Top Pathol 1990; 81: 61-76. Siurala M, Seppala K. Atrophic gastritis as a possible precursor of gastric carcinoma and pernicious anemia. Results of follow-up examinations. Acta Med Scand. 1960 May 5;166:455-74. Stemmermann G, Haenszel W, Locke F. Epidemiologic pathology of gastric ulcer and gastric carcinoma among Japanese in Hawaii . J Natl Cancer Inst. 1977 Jan;58(1):13-20. Stemmermann GN . Intestinal metaplasia of the stomach. A status report. Cancer. 1994 Jul 15;74(2):556-64. Stoicov C , Saffari R, Cai X, Hasyagar C, Houghton J. Molecular biology of gastric cancer: Helicobacter infection and gastric adenocarcinoma: bacterial and host factors responsible for altered growth Molecular biology of gastric cancer: Helicobacter infection and gastric adenocarcinoma: bacterial and host factors responsible for altered growth signal ing. Gene. 2004 Oct 27;341:1-17. signal ing. Gene. 2004 Oct 27;341:1-17. Tahara E . Molecular mechanism of stomach carcinogenesis. J Cancer Res Clin Oncol. 1993;119(5):265-72. Tahara E . Genetic pathways of two types of gastric cancer. IARC Sci Publ. 2004;(157):327-49. Tannenbaum SR, Moran D, Falchuk KR, Correa P, Cuello C. Nitrite stability and nitrosation potential in human gastric juice. Cancer Lett. 1981 Nov;14(2):131-6. Testino G, Cornaggia M, De Iaco F, Gada D. Is it possible with an immunophenotypic study to foresee the oncologic risk of epithelial gastric dysplasia? Hepatogastroenterology. 2002 May-Jun;49(45):601-3. Torrado J , Blasco E, Gutierrez-Hoyos A, Cosme A, Lojendio M, Arenas JI. Lewis systemal terations in gastric carcinogenesis. Cancer. 1990 Oct 15;66(8):1769-74. Torrado J, Correa P, Ruiz B, Bernardi P, Zavala D, Bara J. Lewis antigenal terations in gastric cancer precursors. Gastroenterology. 1992 Feb;102(2):424-30. Torrado J, Correa P, Ruiz B, Zavala D, Bara J. Prospective study of Lewis antigenal terations in the gastric precancerous process. Cancer Epidemiol Biomarkers Prev. 1992 Mar-Apr;1(3):199-205. Torrado J, Plummer M, Vivas J, Garay J, Lopez G, Peraza S, Carillo E, Oliver W, Munoz N. Lewis antigenal terations in a population at high risk of stomach cancer. Cancer Epidemiol Biomarkers Prev. 2000 Jul;9(7):671-4. Yang J, Hu Z, Xu Y, Shen J, Niu J, Hu X, Guo J, Wei Q, Wang X, Shen H. Interleukin-1B gene promoter variants are associated with an increased risk of gastric cancer in a Chinese population. Cancer Lett. 2004 Nov 25;215(2):191-8. Zhang YC. Geographic pathology of gastric dysplasia in China . Semin Surg Oncol. 1994 Mar-Apr;10(2):100-106. Zhang GX , Gu YH, Zhao ZQ, Xu SF, Zhang HJ, Wang HD, Hao B. Coordinate increase of telomerase activity and c-Myc expression in Helicobacter pylori-associated gastric diseases. World J Gastroenterol. 2004 Jun 15;10(12):1759-62. Zullo A, Romiti A, Borrini F, Hassan C, Stella F, Sarcina I, Martini ME, Tomao S, Morini S, Mingazzini P. Alteration of E-cadherin expression in gastric mucosa: role of intestinal metaplasia and Helicobacter pylori infection. Anticancer Res. 2004 May-Jun;24(3a):1603-7.
|
|
|
|
- Oscar Marin (02/10/2005 17:17:38)
- José Miguel Sanz Anquela (02/10/2005 22:51:00)
- Mario Michel Gómez Hernández (04/10/2005 0:51:58)
- Alexandra Gené Heym (04/10/2005 17:03:50)
- Juan Pablo Garcia de la Torre (05/10/2005 8:09:42)
- Carmen López Peña (05/10/2005 20:29:22)
- María Caridad De Armas Fernández (06/10/2005 20:58:18)
- Emilio Mayayo Artal (07/10/2005 19:00:13)
- NATALIA MARTINEZ CASTILLO (10/10/2005 0:22:03)
- Javier Muñoz Moreno (11/10/2005 12:41:45)
- Francisco Javier Flores Figueroa (11/10/2005 20:08:59)
- Liliette Caraballoso García (12/10/2005 4:46:15)
- César Jesús Eras Lévano (12/10/2005 5:19:20)
- hector santiago antunez moncada (12/10/2005 11:24:17)
- ROSA N. RODRÍGUEZ RODRÍGUEZ (13/10/2005 20:22:47)
- ELSIE BEATRIZ PICOTT RANGEL (16/10/2005 10:32:13)
- Juliana Fariña (16/10/2005 13:41:59)
- Mario Miguel Morales Wong (17/10/2005 3:13:14)
- Javier Muñoz Moreno (22/10/2005 14:38:46)
- Carmen Nieves Hernández León (25/10/2005 14:51:22)
- MARÍA ANTONIETA GÓMEZ LAGUNAS (26/10/2005 1:10:41)
- Lizbet María León Herrera (02/11/2005 14:30:36)
- José Miguel Sanz Anquela (10/11/2005 20:31:23)
|
|
|
|
|
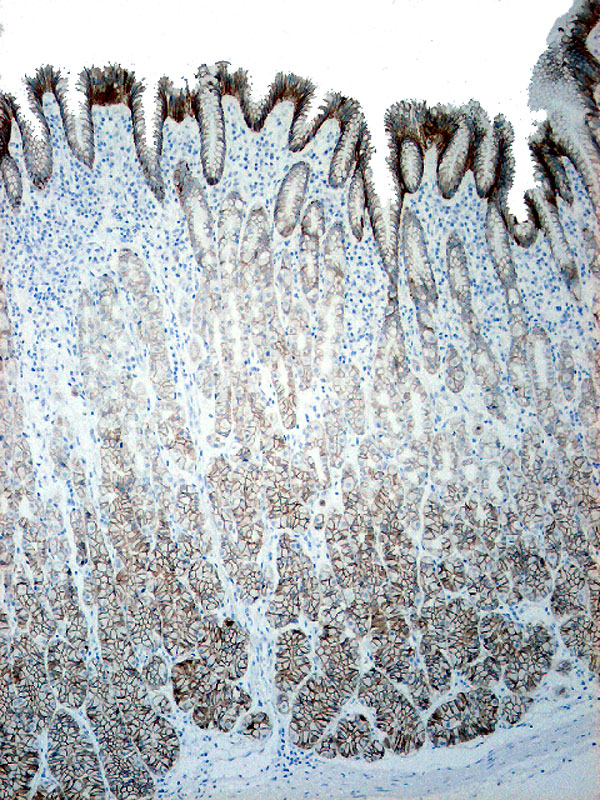
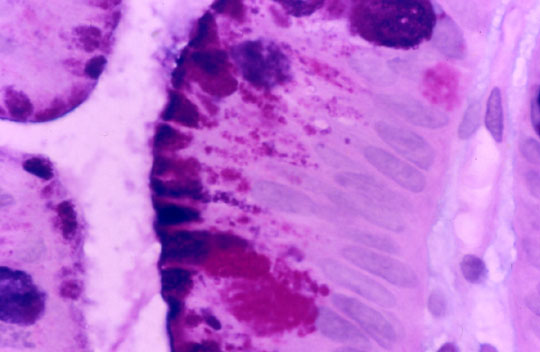
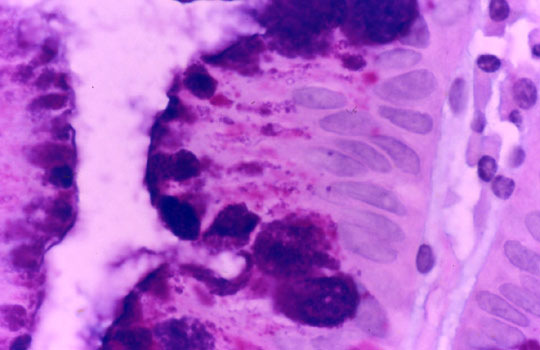
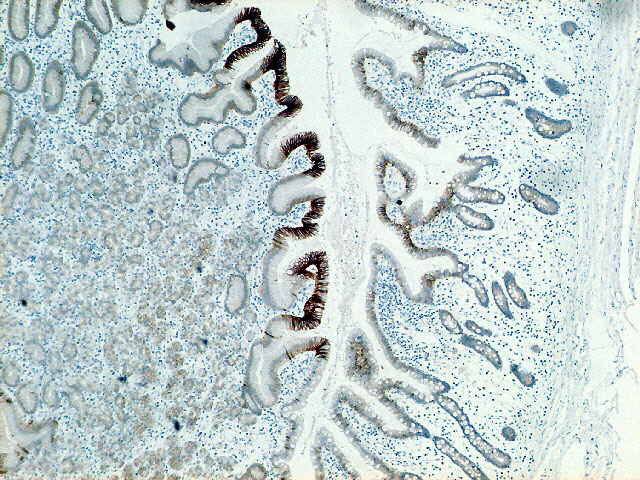 fiogf49gjkf0dDivertículo de Meckel: La elevada expresión de E-cadherina en el epitelio foveolar superficial gástrico de la heterotopia, contrasta con la pérdida de expresión de la mucosa de intestino delgado en el margen derecho de la imagen.">
fiogf49gjkf0dDivertículo de Meckel: La elevada expresión de E-cadherina en el epitelio foveolar superficial gástrico de la heterotopia, contrasta con la pérdida de expresión de la mucosa de intestino delgado en el margen derecho de la imagen.">
Web mantenido y actualizado por el Servicio de informática uclm. Modificado: 16/06/2015 15:10:50