Direccion de contacto
fiogf49gjkf0d nsubiros@infomed.sld.cu
|
|
Evaluación histológica del efecto de la Eritropoyetina vía intranasal, sobre la muerte neuronal retardada en gerbos sometidos a isquemia cerebral transitoria. Estudio preliminar.
Nelvys Subirós Martínez*, Julio César García Rodríguez**, Bárbara González Navarro**, Iliana Sosa Testé**, Jorge Daniel García Salman*
* Instituto de Neurología y Neurocirugía CUBA
** Centro Nacional para la Producción de Animales de Laboratorio CUBA
|
|
|
Resumen
fiogf49gjkf0d La eritropoyetina (EPO) es una citocina, comúnmente conocida como estimulador hormonal del eritropoyesis, que actúa como un factor de crecimiento e inhibidor de la apoptosis. Recientemente se ha demostrado la expresión del sistema EPO/Receptor de EPO en el cerebro, específicamente en áreas como el hipocampo, donde se localizan neuronas muy vulnerables al insulto isquémico. Estudios recientes han demostrado que la EPO humana recombinante puede proporcionar neuroprotección en diferentes modelos de daño cerebral. Nuestro objetivo fue evaluar de forma preliminar el efecto de la EPO-CIM en el modelo de isquemia transitoria en gerbos de Mongolia, por oclusión de las arterias carótidas comunes. Se establecieron cinco grupos experimentales: animales no lesionados, tres grupos de animales con tiempo de oclusión de 5, 8 y 10 minutos, y un grupo de 10 minutos de oclusión con 120µg/día de EPO-CIM. Luego de 7 días los animales se perfundieron con formol neutro al 10%, se extrajo el cerebro cuidadosamente y las muestras se procesaron para microscopía óptica. Se trabajó con cortes de 3 micras, teñidas con H&E. Con el objetivo de 40x, se contó el número de neuronas del estrato piramidal de CA1, en ambos hemisferios y se calculó la densidad de células/mm. Todos los grupos lesionados sin tratamiento mostraron una densidad de neuronas piramidales significativamente más baja que los animales controles sanos y los tratados con EPO; no se encontraron diferencias significativas entre estos dos grupos demostrando el efecto neuroprotector de la EPO-CIM.
|
|
|
Introduccion
fiogf49gjkf0d La isquemia cerebral global, junto con la isquemia cerebral focal, son enfermedades de la población humana, causantes de muerte y discapacidad. En la isquemia cerebral global se produce un cese completo del flujo sanguíneo cerebral, lo que provoca daño neuronal en áreas selectivamente vulnerables (Traystman, 2003).
La neuroprotección es una estrategia de tratamiento para enfermedades del Sistema Nervioso Central (SNC) de diferentes orígenes fisiopatológicos, como infarto, neurotrauma, enfermedades neuroinflamatorias y neurodegenerativas; y su objetivo es prevenir u oponerse a la pérdida neuronal (Sirén y Ehrenreich, 2001a). Hasta el momento, han sido muchos los agentes neuroprotectores que han tenido resultados muy prometedores en estudios con modelos animales de daño neurológico, y sin embargo los resultados de los ensayos clínicos se muestran decepcionantes, pues ninguno ha mostrado beneficios inequívocos en estudios de fase III, además de la toxicidad manifiesta en humanos (Sirén y Ehrenreich, 2001a). La excepción ha sido el activador tisular del plasminógeno, aunque tiene una utilidad limitada, por lo que todavía se continúa en la búsqueda de terapias aplicables de una forma más general para el tratamiento de la isquemia aguda y el infarto hemorrágico (Ovbiagele, 2003).
Actualmente, varios grupos de investigadores trabajan en un novedoso agente neuroprotector: la eritropoyetina humana recombinante (EPOhr). Se ha comprobado que la EPO y su receptor se expresan en el tejido cerebral, además de que su expresión aumenta durante la isquemia cerebral, hipoxia y anemia, lo que sugiere su participación en un sistema neuroprotector endógeno en el cerebro de mamíferos (Marti et al, 2000). La efectividad de la EPOhr como neuroprotector se ha probado en múltiples modelos de daño del sistema nervioso (en ratón, rata, gerbo y conejo), incluyendo isquemia cerebral focal y global, donde se ha visto que reduce la muerte neuronal (Bernaudin et al., 1999; Brines et al., 2000; Calapai et al, 2000; Catania et al., 2002; Erbayraktar et al., 2003; Sadamoto et al., 1998; Sakanaka et al., 1998; Sirén et al, 2001b; Sirén et al., 2001c).
Aunque todavía se investiga el mecanismo neuroprotector de la EPO, se ha planteado que potencia mecanismos antiapoptóticos, antioxidantes, además de tener acción neurotrófica, anti-inflamatoria, angiogénica y moduladora de la actividad neuronal (Ehrenreich, 2004).
En la literatura se ha tratado también el tema de la entrada de la molécula al SNC. Se ha reportado el uso de inyecciones intracerebroventriculares de EPOhr , con las que se ha obtenido una reducción del infarto en ratones con isquemia cerebral focal (Bernaudin et al., 1999; Sadamoto et al., 1998, Sakanaka et al., 1998), aunque se comprende lo impracticable del método en la clínica. Actualmente se emplea la vía sistémica en animales, pues hay evidencias de que la EPOhr cruza la barrera hemato-encefálica y protege de manera efectiva el daño isquémico cerebral (Brines et al., 2000). Recientemente se realizó un ensayo clínico, en el que se administraron altas dosis de EPOhr por vía endovenosa como tratamiento agudo en la isquemia cerebral. En los resultados se obtuvo un mejoramiento significativo de los pacientes infartados, y una buena tolerancia del producto (Ehrenreich et al., 2002).
Sin embargo, muchas situaciones clínicas probablemente necesitarían múltiples dosis de EPOhr, lo cual podría conllevar a un aumento no deseado de la masa de células eritroides (Wiessner et al, 2001). La variante de una administración periférica de una molécula de EPOhr con bajo contenido de ácido siálico (Erbayraktar et al., 2003), tiene la desventaja que ésta resultaría fácilmente degradable a nivel hepático.
Una novedosa vía para hacer llegar la EPOhr al SNC es a través de la cavidad nasal. En la parte superior de la misma se encuentran las terminales nerviosas responsables de conducir la información del olor a través de la placa cribiforme (Chien et al., 1989). Por los orificios de la placa cribiforme pasan los haces de cordones nerviosos que constituyen el tracto olfativo del SNC, que se extiende desde la base del cerebro a diferentes regiones subcorticales. En experimentos anteriores de nuestro grupo de trabajo, se ha comprobado la entrada de la EPOhr al SNC por esta vía, en un tiempo menor que al ser administrada sistémicamente (resultados no publicados).
Para este trabajo, se trabajó con el modelo de isquemia global en el gerbo de Mongolia. En estos animales, puede inducirse una isquemia cerebral global por oclusión bilateral de las arterias carótidas comunes (ACC), ya que en su árbol vascular no aparece la arteria comunicante posterior, aislando el sistema arterial carotídeo del vertebrobasilar (Levine and Sohn 1969). Durante la oclusión el flujo sanguíneo cerebral se reduce casi a cero en la mayoría de los animales (Crockard et al., 1980). En este modelo, una oclusión de 5 minutos resulta en muerte de las neuronas de CA1 del hipocampo (Kirino, 1982).
El objetivo de este estudio es evaluar el efecto neuroprotector de la EPOhr-CIM administrada por vía nasal en el modelo de isquemia cerebral global en gerbos de Mongolia, así como estudiar los efectos de diferentes tiempos de oclusión de las ACC en el encéfalo.
|
|
|
Material y Métodos
fiogf49gjkf0d Se emplearon gerbos de Mongolia machos (n=33), con un peso corporal entre 60 y 70 gramos, obtenidos del Centro Nacional para la Producción de Animales de Laboratorio (CENPALAB, Cuba). Los animales se alojaron de forma individual, con libre acceso al agua y al alimento. Se estableció un ciclo de 12 horas alternas de luz oscuridad durante todo el período experimental.
La EPO fue suministrada por el Centro de Inmunología Molecular (CIM, Habana, Cuba), con una concentración de 1mg/ml, y pH neutro.
Grupos experimentales
Se establecieron cinco grupos experimentales (ver tabla).
|
Grupo |
Lesión |
Tiempo de oclusión |
N |
Tratamiento |
|
1 |
Falsa lesión |
- |
9 |
- |
|
2 |
Isquemia - reperfusión |
5 min |
5 |
- |
|
3 |
Isquemia reperfusión |
8 min |
5 |
- |
|
4 |
Isquemia reperfusión |
10 min |
5 |
- |
|
5 |
Isquemia reperfusión |
10 min |
9 |
EPOhr-CIM |
El grupo tratado con EPOhr se administró por vía nasal, a las 0, 1, 2 y 3 horas después de la operación, y luego 2 veces diarias durante 3 días. Cada aplicación fue de 120 microgramos de EPO /animal.
Cirugía
Para la operación, los animales se anestesiaron por vía intraperitoneal, empleando como pre-anestésico diazepán (5.0 mg/kg.) y como anestésico ketamina (47 mg/kg.) y 0,02 mg/kg de atropina. Se realizó una incisión longitudinal ventral, se localizaron las ACC de ambos lados, cuidando de no lesionar el tronco vagosimpático. Luego se procedió a ocluirlas sin interrupción, durante el tiempo establecido para cada animal. Tras la liberación de las arterias se comprobó el restablecimiento del flujo sanguíneo y se procedió a suturar la herida. Durante el proceso quirúrgico y la recuperación de la anestesia, la temperatura corporal de los animales se mantuvo a 37 ± 0.1oC utilizando un sistema de registro y control de temperatura (Barnand, USA).
En los animales falsamente lesionados se realizó el mismo procedimiento quirúrgico, excepto que a estos no se les ocluyeron las ACC.
Histología
Al séptimo día de la operación, los gerbos fueron perfundidos bajo anestesia con formalina neutra tamponada al 10%. Se extrajeron los encéfalos cuidadosamente y se mantuvieron en la solución fijadora por varios días.
Se tomaron 2 secciones coronales de 2mm de grosor aproximadamente, una de ellas se ubicó siempre en la región de 1.4 a 2.0 mm posterior a bregma, según el atlas estereotáxico (Loskota et al., 1974), para asegurarnos de trabajar hipocampo dorsal. La otra sección coronal correspondió a la región anterior. Estas muestras se incluyeron en parafina y se obtuvieron cortes de 4-5 micras de espesor, que fueron teñidos con hematoxilina - eosina.
Las láminas fueron analizadas por un patólogo especialista, registrándose la descripción cualitativa de cada animal. Con el objetivo de calcular la densidad de células piramidales/mm (Kirino, 1982) se tomaron imágenes del estrato de CA1 del hipocampo de ambos hemisferios con el objetivo de 40x, en las que se contó el número de neuronas aparentemente sanas. Se tuvieron en cuenta aquellas neuronas con núcleo redondeado, cromatina laxa, de tamaño relativamente grande y no eosinofílicas. En todos los casos se analizó una distancia igual a 1.6 mm, garantizada con el sistema cubano para morfometría de imágenes DIGIPAT, versión 3.3 sobre Windows. El conteo se realizó sin conocimiento previo de la identificación de las láminas.
Procesamiento estadístico: Los grupos se compararon mediante el Análisis de Varianza de una vía (p<0.05), seguido de una prueba Tukey. Se empleó el paquete estadístico SPSS, versión 8.0.
|
|
|
Resultados
fiogf49gjkf0d El grupo de animales falsamente lesionados no mostró daño. En todos los grupos se observaron neuronas oscuras de forma poco frecuente, lo cual es característico del tejido fijado en aldehído.
En los tres grupos de isquemia no tratada se observó una pérdida casi total de neuronas piramidales del sector CA1 del hipocampo, como indican los valores de densidad de neuronas de estos grupos (Tabla). Este daño estuvo caracterizado por contracción y eosinofilia de los cuerpos neuronales, con núcleos en cariorresis y cariolisis (Fig. 1). Cualitativamente no se observaron grandes diferencias entre los grupos con diferentes tiempos de oclusión. En algunos casos la pérdida neuronal no alcanza el mismo grado en ambos hemisferios. Aunque poco frecuentes también se observaron estructuras que se asemejan a cuerpos apoptóticos (Fig. 1).
En el grupo tratado con EPOhr la mayoría de los animales no tenía daño en el sector CA1, o este era un daño ligero (Fig. 2). En tres de los animales sí se encontró un daño severo.
En el grupo de 10 minutos de oclusión de las ACC, también se ven afectadas otras áreas, fundamentalmente en la sustancia gris. Estas afectaciones se localizan principalmente en núcleos del tálamo, como en los núcleos postero-medial ventral y ventro medial, y en núcleos del hipotálamo. Las lesiones se caracterizan por desmielinización, gliosis con proliferación de corpúsculos granulo grasosos, algunas mitosis y una ligera espongiosis del neuropilo (Fig. 3). Estas lesiones se presentan de forma bilateral.
La comparación entre grupos de la densidad de células piramidales en CA1 a los 7 días del daño mostró que el grupo tratado con EPOhr es estadísticamente igual al grupo falsamente lesionado y además significativamente mayor que los grupos sin tratamiento (Tabla).
|
Grupo |
N |
Piramidales/mm (X) |
SD |
|
Falso lesionado |
9 |
259 a |
34 |
|
Lesión 5 minutos |
5 |
39 b |
53 |
|
Lesión 8 minutos |
5 |
22 b |
15 |
|
Lesión 10 minutos |
5 |
15 b |
12 |
|
Lesionado 10 minutos + EPOhr |
9 |
171 a |
115 |
Densidad de neuronas piramidales de CA1. N: cantidad de animales por grupo. X: Media. SD: Desviación estándar. Letras diferentes indican diferencias significativas (p<0.05)
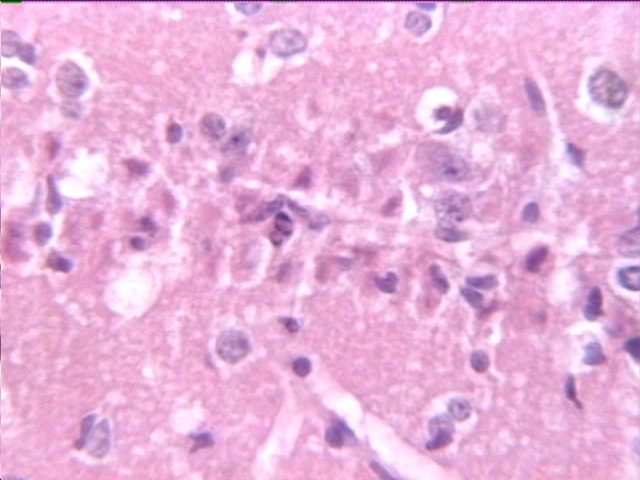 fiogf49gjkf0dFigura 1. Lesión en CA1 en isquemia por 10 minutos. Necrosis de neuronas y presencia de cuerpos apoptóticos (flecha). H-E. x40."> fiogf49gjkf0dFigura 1. Lesión en CA1 en isquemia por 10 minutos. Necrosis de neuronas y presencia de cuerpos apoptóticos (flecha). H-E. x40.">
isquemia en CA1 - fiogf49gjkf0d Figura 1. Lesión en CA1 en isquemia por 10 minutos. Necrosis de neuronas y presencia de cuerpos apoptóticos (flecha). H-E. x40.
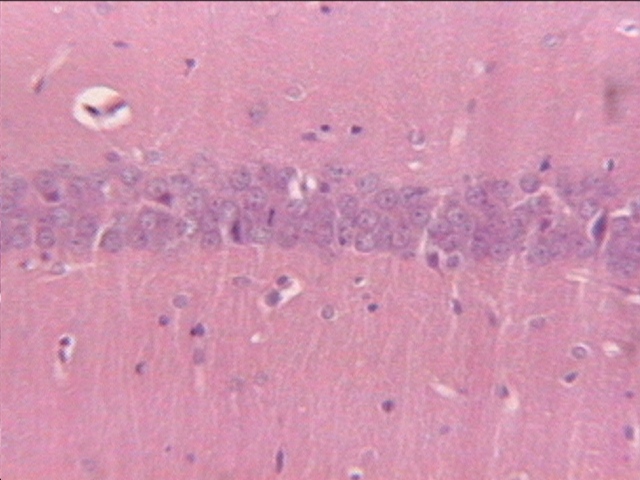 fiogf49gjkf0digura 2. Área de CA1 de animal en isquemia por 10 minutos y tratado con EPOhr. Preservación de las neuronas. H-E. x20."> fiogf49gjkf0digura 2. Área de CA1 de animal en isquemia por 10 minutos y tratado con EPOhr. Preservación de las neuronas. H-E. x20.">
- fiogf49gjkf0d igura 2. Área de CA1 de animal en isquemia por 10 minutos y tratado con EPOhr. Preservación de las neuronas. H-E. x20.
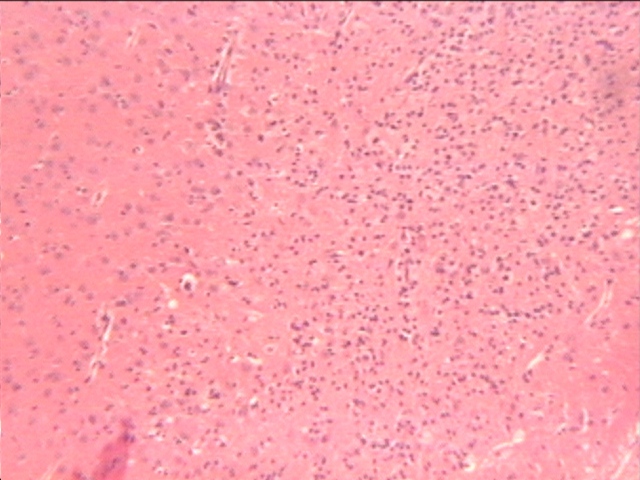 fiogf49gjkf0dFigura 3. Fotografías tomada del tálamo de un animal lesionado durante 10 minutos. Foco de gliosis y espongiosis del neuropilo. H-E (x10)."> fiogf49gjkf0dFigura 3. Fotografías tomada del tálamo de un animal lesionado durante 10 minutos. Foco de gliosis y espongiosis del neuropilo. H-E (x10).">
- fiogf49gjkf0d Figura 3. Fotografías tomada del tálamo de un animal lesionado durante 10 minutos. Foco de gliosis y espongiosis del neuropilo. H-E (x10).
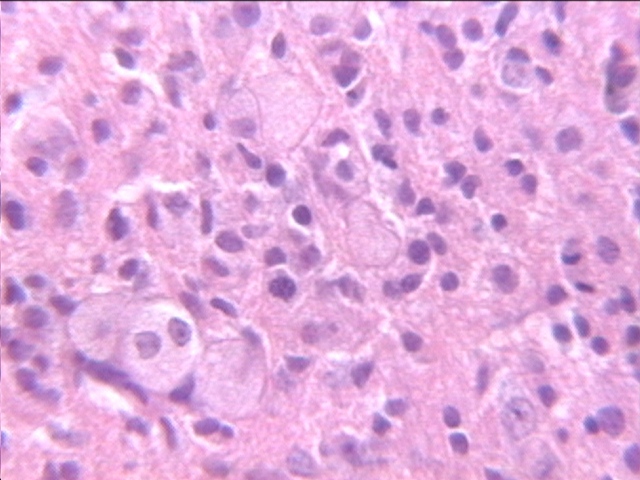 fiogf49gjkf0dFigura 4. Zona de gliosis donde concomitan los corpúsculos granulo grasosos. H-E. (x40)."> fiogf49gjkf0dFigura 4. Zona de gliosis donde concomitan los corpúsculos granulo grasosos. H-E. (x40).">
- fiogf49gjkf0d Figura 4. Zona de gliosis donde concomitan los corpúsculos granulo grasosos. H-E. (x40).
|
|
|
Discusión
fiogf49gjkf0d En la literatura pueden encontrarse numerosos estudios, tanto in vivo como in vitro, que demuestran los efectos neuroprotectores de la EPOhr durante la isquemia cerebral focal y global (Bernaudin et al., 1999,Brines et al., 2000; Calapai et al., 2000; Catania et al., 2002; Erbayraktar et al., 2003; Sadamoto et al., 1998), la encefalitis autoinmune experimental, los ataques inducidos por ácido kaínico, daño cerebral traumático (Brines et al., 2000), daño cerebral hipóxico isquémico neonatal (Kumral et al., 2003), entre otros (Genc et al., 2004). Los primeros estudios in vivo administraron la EPOhr directamente en los ventrículos cerebrales de los animales con el objetivo de hacer llegar la molécula al tejido sin que tenga que cruzar la barrera hematoencefálica (BHE) (Bernaudin et al., 1999, Sadamoto et al., 1998, Sakanaka et al., 1998). La posibilidad de que la EPO pueda pasar la BHE es fundamental para su aplicación como agente neuroterapéutico en la práctica clínica ya que la administración intratecal es impracticable. Brines et al., (2000) demostraron que la EPO administrada sistémicamente cruza la BHE y reduce el daño tisular de forma significativa en un modelo animal de insulto isquémico. Otros estudios han utilizado esta vía para hacer llegar la EPOhr al cerebro, de hecho el primer ensayo clínico demostró un mejoramiento significativo de la evolución de los pacientes (Ehrenreich et al., 2002).
Nuestros resultados, coincidiendo con otros trabajos ya publicados (Sakanaka et al., 1998; Catania et al., 2002), muestran que el tratamiento con EPOhr aumentó significativamente el número de células aparentemente sanas. La densidad de neuronas del grupo de animales tratados con EPOhr, el cual no difiere del grupo control (falsamente lesionado), evidencia que la EPOhr protege a las neuronas piramidales de CA1 de un daño isquémico letal como el que se produce en el modelo de isquemia cerebral global en el gerbo, lo cual ya ha sido demostrado anteriormente (Sakanaka et al., 1998). Este efecto protector, constituye una evidencia del paso de la EPOhr a través de la cavidad nasal.
Hacer llegar la EPOhr al cerebro a través de la vía nasal permite evadir la barrera hematoencefálica ya que su paso podría estar mediado por un mecanismo general y rápido como es la difusión a través del mucus, y ulterior penetración a través de discontinuidades que se presentan en regiones del epitelio olfativo, en particular en la zona de la lámina cribiforme, entrando en contacto con el líquido cefalorraquídeo, por medio del cual podría ser ampliamente diseminada por el SNC (Frey et al., 1997). Además, esta vía de administración podría ser mucho más segura y rápida, además de no invasiva, que la vía endovenosa, utilizada para hacer llegar medicamentos al cerebro dañado. Se ha reportado el transporte de sustancias hacia el cerebro por la vía nasal, de una forma rápida (Sakane et al., 2000), de manera que esta vía podría ser utilizada para el tratamiento profiláctico y/o terapéutico con EPOhr ante distintas injurias agudas o crónicas.
En cuanto a los mecanismos moleculares por los que la EPOhr media sus efectos protectores en el SNC, estos aún no se han descifrado completamente. Por analogía con las células precursoras eritroides en la médula ósea, donde la EPO promueve la viabilidad celular inhibiendo la apoptosis (Koury y Bondurant, 1990), se ha especulado que en las células neuronales y endoteliales operan mecanismos similares (Marti, 2004).
Las vías de señales moleculares que median el efecto neuroprotector de la EPO en las células comienzan por la activación del Receptor de la EPO, que en forma de homodímero permite la autofosforilación de la Janus tirosin kinasa 2 (JAK2). Esta a su vez conlleva a la fosforilación de varias cascadas de señales, incluyendo la activación de la proteína kinasa activada por mitógeno Ras (MAPK), la fosfatidil inositol kinasa (PI(3)K) y el factor de transcripción Stat5 (signal transducers and activators of transcription) (Marti, 2004). En las células diana, el efecto neto de la estimulación del Receptor de EPO es la proliferación, la inhibición de la apoptosis, y en el caso de los eritroblastos, la diferenciación (Yoshimura and Misawa, 1998).
Sakanaka et al., (1998) reportaron que una infusión intracerebral del Receptor de EPO en forma soluble neutraliza la EPO endógena y resulta en una muerte neuronal por apoptosis masiva en CA1 después de una isquemia global no letal, lo que evidencia una acción anti-apoptótica intrínseca. Estudios recientes también dan evidencias de este efecto, cuando la molécula es administrada por vía intraperitoneal (Sirén et al., 2001c). Se ha estudiado además el aumento de la expresión de genes anti-apoptóticos como el bcl-xL, la disminución de la expresión de genes pro-apoptóticos como Bak en células PC12 (Renzi et al., 2002) y la inhibición de la actividad de las caspasas 8, 1 y 3, relacionadas con la liberación de citocromo c de la mitocondria (Chong et al., 2003), entre otros hallazgos (Genc et al., 2004 y Marti, 2004). Todo lo anterior sugiere que sus efectos pueden involucrar el control del balance de expresión de moléculas pro- y anti-apoptóticas (Genc et al., 2004). Se plantea que la EPO tiene también actividad neurotrófica, lo que implica un efecto de mayor latencia que la inhibición de la apoptosis (Sirén y Ehrenreich, 2001a)
Por otro lado, el modelo animal de isquemia reperfusión en el gerbo de Mongolia implica un estrés oxidativo, cuyo daño al DNA y otras moléculas provoca muerte celular por necrosis o apoptosis (Love, 1999, Warner et al., 2004). Este es otra vía en la que la intervención de la EPO puede contribuir a un efecto citoprotector. Se ha propuesto que esta molécula suprime la formación de radicales libres mediados por el óxido nítrico o actúa como antagonista de su toxicidad (Sakanaka et al., 1998). Su acción antioxidante también se sustenta en el reporte de que la EPO protege contra el estrés oxidativo a través de la reducción de la peroxidación lipídica y el restablecimiento de las actividades de la catalasa citosólica y la Glutatión peroxidasa en eritrocitos (Chattopadhyay et al., 2000).
En la isquemia cerebral global hay ausencia de flujo sanguíneo en el cerebro, lo que causa un daño neuronal en áreas selectivamente vulnerables (Traystman, 2003). Entre las respuestas cerebrales específicas al paro circulatorio, se han diferenciado dos categorías de lesiones. En primer lugar está la muerte neuronal retardada en regiones selectivamente vulnerables, de forma notable en el hilus y el sector CA del hipocampo, el estriado dorsal y el núcleo reticular del tálamo. Estas lesiones se producen consistentemente por episodios breves de isquemia global. En la segunda categoría se agrupan lesiones necróticas en otras partes del cerebro, las cuales dependen de la duración, amplitud y temperatura de la isquemia, y también del estado de reperfusión post isquémica (Hossman, 1998).
En el modelo de isquemia cerebral global en el gerbo de Mongolia, la oclusión bilateral de 5 minutos de las ACC resulta en daño y muerte de las neuronas hipocampales de CA1 (Kirino, 1982). Nuestros hallazgos cualitativos, analizados en función de la duración de la isquemia, se corresponden con lo planteado anteriormente. El tiempo de oclusión de las ACC por 5 minutos fue suficiente para provocar un daño isquémico cerebral, que a los 7 días de la lesión se expresó en la muerte selectiva de la mayoría de las neuronas piramidales de CA1 del hipocampo y las características morfológicas de este tipo de muerte se corresponden en su gran mayoría con la necrosis (Colbourne et al., 1999). En la actualidad existe un consenso de que también la apoptosis es un mecanismo de muerte que ocurre en condiciones de isquemia menos severa, aunque han existido opiniones contrarias (Colbourne et al., 1999). De esta manera, los cuerpos encontrados en varios de nuestros animales, y que recuerdan la morfología de cuerpos apoptóticos, necesitan comprobación por técnicas como la microscopía electrónica.
Las lesiones que encontramos al aumentar la duración de la isquemia (isquemia durante 10 minutos) se corresponden con la segunda categoría de respuestas específicas del cerebro, como plantea Hossmann (1998). La gliosis es una respuesta de las células neurogliales en el SNC a muchas formas de injuria, entre ellas, los procesos isquémicos. Un área con gliosis es hipercelular a causa de la hipertrofia de células gliales, la proliferación celular (más característica de la microglia que de la astroglia) o ambos fenómenos (Summers et al, 1995). En conclusión, nuestro experimento pudo demostrar que a partir del paso de la EPOhr a través de la cavidad nasal, esta ejerce un efecto neuroprotector sobre las neuronas del hipocampo. Además, se pudo establecer que con un tiempo de oclusión bilateral de la ACC de 5 minutos el modelo utilizado logra resultados óptimos.
|
|
|
Bibliografía
· Bernaudin M, Marti HH, Roussel S, Divoux D, Nouvelot A, MacKenzie ET, Petit E. (1999) A potential role for erythropoietin in focal permanent cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 19: 643 651.
· Brines ML, Ghezzi P, Keenan S, Agnello D, de Lanerolle NC, Cerami C, Itri LM, Cerami A. (2000) Erythropoietin crosses the bloodbrain barrier to protect against experimental brain injury. Proc. Natl. Acad. Sci. U. S. A. 97: 10526 10531.
· Calapai G, Marciano MC, Corica F, Allegra A, Parisi A, Frisina N, Caputi AP, Buemi M. (2000) Erythropoietin protects against brain ischemic injury by inhibition of nitric oxide formation. Eur. J. Pharmacol. 401: 349 356.
· Catania MA, Marciano MC, Parisi A, Sturiale A, Buemi M, Grasso G, Squadrito F, Caputi AP, Calapai G. (2002) Erythropoietin prevents cognition impairment induced by transient brain ischemia in gerbils. Eur. J. Pharmacol. 437: 147 150
- Chattopadhyay A, Choudhury TD, Bandyopadhyay D, Datta AG. (2000) Protective effect of erythropoietin on the oxidative damage of erythrocyte membrane by hydroxyl radical. Biochem. Pharmacol. 59: 419 425.
· Chien Y.W., Su S.E.K., Chang S.F. (1989) Nasal Systemic drug delivery. Drugs and the pharmaceutical science. Marcel Dekker, INC. New York, 2da Ed.
· Chong ZZ, Kang JQ, Maiese K. (2003) Erythropoietin fosters both intrinsic and extrinsic neuronal protection through modulation of microglia, Akt1, Bad, and caspase-mediated pathways. Br. J. Phar-macol. 138: 1107 1118.
· Colbourne F, Sutherland GR, Auer RN. (1999) Electron Microscopic Evidence against Apoptosis as the Mechanism of Neuronal Death in Global Ischemia. The Journal of Neuroscience. 19(11): 4200-4210
- Crockard A, Ianotti F, Hunstock AT, Smith RD, Harris RJ, Symon L. (1980) Cerebral blood flow and edema following carotid occlusion in the gerbil. Stroke 11: 494-498.
· Ehrenreich H. (2004) A Boost for Translational Neuroscience. Science. 305: 184-185.
- Ehrenreich H, Hasselblatt M, Dembowski C, Cepek L, Lewczuk P, Stiefel M, Rustenbeck HH, Breiter N, Jacob S, Knerlich F, Bohn M, Poser W, Ruther E, Kochen M, Gefeller O, Gleiter C, Wessel TC, De Ryck M, Itri L, Prange H, Cerami A, Brines M, Siren AL. (2002) Erythropoietin therapy for acute stroke is both safe and beneficial. Mol. Med. 8: 495 505.
· Erbayraktar S, Grasso G, Sfacteria A, Xie QW, Coleman T, Kreilgaard M, Torup L, Sager T, Erbayraktar Z, Gokmen N, Yilmaz O, Ghezzi P, Villa P, Fratelli M, Casagrande S, Leist M, Helboe L, Gerwein J, Christensen S, Geist MA, Pedersen LO, Cerami- Hand C, Wuerth JP, Cerami A, Brines M. (2003) Asialoerythropoietin is a nonerythropoietic cytokine with broad neuroprotective activity in vivo. Proc. Natl. Acad. Sci. U. S. A. 100: 6741 6746.
· Frey HW, Liu J, Chen X, Thoner RG, Fawcett JR, Ala TA, Rahman YE. (1997) Delivery of 125 I-NGF to the brain via the olfatory Route. Drug Delivery. 4: 87-92.
· Genc S, Koroglu TF, Genc K. (2004) Erythropoietin and the nervous system. Brain Research 1000: 1931
· Hossmann KA. (1998) Experimental models for the investigation of brain ischemia. Cardiovascular Research 39: 106120.
- Kirino T. (1982) Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res 239: 57-69.
· Koury, M. J. and Bondurant, M. C. (1990) Erythropoietin retards DNA breakdown and prevents programmed death in erythroid progenitor cells. Science 248: 378 -381
- Kumral A, Ozer E, Yilmaz O, Akhisaroglu M, Gokmen N, Duman N, Ulukus C, Genc S, Ozkan H. (2003) Neuroprotective effect of erythropoietin on hypoxic ischemic brain injury in neonatal rats. Biol. Neonate 83: 224 228.
- Levine S, Sohn D. (1969) Cerebral ischemia in infarct and adult gerbils: Relation to incomplete Circle of Willis. Arch Pathol 87: 315-317.
· Loskota WJ, Lomax P, Verity MA. (1974) A stereotaxic atlas of the Mongolian gerbil brain (Meriones unguiculatus). Ann Arbor Science Publishers Inc.
- Love S. (1999) Oxidative Stress in Brain Ischemia. Brain Pathology 9: 119-131
· Marti H, Bernaudin M, Petit E, Bauer C. (2000) Neuroprotection and Angiogenesis: Dual role of erythropoietin in brain ischemia. News of Physiology Science. 15: 225-229.
· Marti HH. (2004) Erythropoietin and the hypoxic brain. Journal of Experimental Biology 207: 3233-3242
· Ovbiagele B, Kidwell C, Starkman S y Saver J. (2003) Potential Role of Neuroprotective Agents in the Treatment of Patients with Acute Ischemic Stroke. Current Treatment Options in Neurology. 5: 367375.
· Renzi MJ, Farrell FX, Bittner A, Galindo JE, Morton M, Trinh H, Jolliffe LK. (2002) Erythropoietin induces changes in gene expression in PC12 cells. Brain Res. 104: 8695.
· Sadamoto Y, Igase K, Sakanaka M, Sato K, Otsuka H, Sakaki S, Masuda S, Sasaki R. (1998) Erythropoietin prevents place navigation disability and cortical infarction in rats with permanent occlusion of the middle cerebral artery. Biochem. Biophys. Res. Commun. 253: 26 32.
· Sakanaka M, Wen TC, Matsuda S, Masuda S, Morishita E, Nagao M, Sasaki R. (1998) In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc. Natl. Acad. Sci. U. S. A. 95: 4635 4640.
· Sakane, T., Akizuki, M., Yoshida M., Yamashita, S., Nadai, T., Hashida M., and Sezaki H. (2000) Transport of cephalexin to the cerebrospinal fluid directly from the nasal cavity. J. Pharm. Pharmacol. 43: 449-451.
· Sirén AL y Ehrenreich H. (2001a) EPO - a novel concept for neuroprotection. Eur Arch Psychiatry Clin Neurosci. 251: 179184.
· Sirén AL, Knerlich F, Poser W, Gleiter CH, Bruck W, Ehrenreich H. (2001b) Erytropoietin and Erytropoietin receptor in human ischemic/hypoxic brain. Acta Neuropathol. (Berl.) 101: 271-276
· Sirén AL, Fratelli M, Brines M, Goemans C, Casagrande S, Lewczuk P, Keenan S, Gleiter C, Pasquali C, Capobianco A, et al. (2001c). Erythropoietin prevents neuronal apoptosis after cerebral ischemia and metabolic stress. Proc. Natl. Acad. Sci. USA 98: 4044 -4049.
- Summers BA, Cummings JF, de Lahunta A. (1995) Veterinary Neuropathology. Mosby-Year Book, USA. pp 527.
· Traystman RJ. (2003) Animal Models of Focal and Global Cerebral Ischemia. ILAR Journal. 44 (2): 85-95.
· Warner DS, Sheng H, Batini-Haberle I. (2004) Oxidants, antioxidants and the ischemic brain. Journal of Experimental Biology 207: 3221-3231
· Wiessner C, Allegrini PR, Ekatodramis D, Jewell UR, Stallmach T, Gassmann M. (2001) Increased cerebral infarct volumes in polyglobulic mice overexpressing erythropoietin. J. Cereb. Blood Flow Metab. 21: 857864.
· Yoshimura, H. Misawa. (1998) Physiology and function of the erythropoietin receptor. Curr. Opin. Hematol. 5: 171 176.
|
|
|
Comentarios
- Emilio Mayayo Artal (06/10/2005 19:42:49)
Magnífico trabajo experimental que si puede traducir sus resultados a la clínica, nos dará una luz a muchos problemas para los neurólogos, pero sobre todo a muchos pacientes. Enhorabuena por el planteamiento y desarrollo. Emilio.
|
|
|
|
|

 Comunciación libre
Comunciación libre