

|
Sarcoma histiocítico: una neoplasia agresiva con pobre respuesta a la quimioterapia José Luis Villar Rodríguez*, Luis de la Cruz Merino**, José Ibáñez Martínez***, Antonio García Escudero***, Mario Díaz Delgado***, Alejandro Antúnez Infante***, Alicia Hernández Amate***, Ricardo González Cámpora*** |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
El sarcoma histiocítico (SH) (llamado también linfoma histiocítico verdadero) es una neoplasia muy poco frecuente y de estirpe controvertida que puede plantear dificultades en el diagnóstico histopatológico. Por otra parte, su pobre respuesta al tratamiento oncológico y la falta de publicaciones con series amplias de casos lo convierten en un verdadero reto terapéutico. Presentamos para discusión el caso de una mujer de 36 años con un síndrome antifosfolípido, diagnosticada tras una biopsia ganglionar de un SH. Histológicamente el ganglio tenía borrada su arquitectura debido a una población de células grandes, no cohesivas, con un amplio citoplasma eosinófilo, núcleo excéntrico de cromatina vesicular y nucléolo evidente. Entre ellas se advertía una matriz mixoide, abundantes estructuras vasculares y numerosos polinucleares neutrófilos. Las mitosis eran frecuentes y muy atípicas. El estudio inmunohistoquímico demostró que las células neoplásicas eran fuertemente positivas para CD68 y lisozima, y negativas para marcadores linfoides B y T, citoqueratinas, AME, proteina S-100, HMB45, mieloperoxidasa, CD1a y marcadores de células dendríticas. Durante dos años y hasta su fallecimiento la paciente recibió radioterapia local (40 Gy) y numerosos ciclos de distintos protocolos quimioterápicos (CHOP, MINE+G, taxol+topotecan, CVP y, finalmente, fludarabina), a pesar de lo cual desarrolló enfermedad progresiva con afectación de las partes blandas de los miembros inferiores y de distintos territorios ganglionares.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
El sarcoma histiocítico (SH) (llamado antes linfoma histiocítico verdadero) es una neoplasia muy poco frecuente y de estirpe controvertida que puede plantear dificultades en el diagnóstico histopatológico. Por otra parte, su pobre respuesta al tratamiento oncológico y la falta de publicaciones con series amplias de casos lo convierten en un verdadero reto terapéutico.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Mujer de 36 años con un síndrome antifosfolípido que consultó por una adenopatía inguinal derecha de 4 x 3 cm. En la TAC se apreciaron adenomegalias inguinales e ilíacas derechas, y en la PAAF del ganglio se obtuvieron células neoplásicas pleomórficas. El estudio histológico reveló una arquitectura ganglionar borrada (Fig 1) a expensas de una población de células grandes, no cohesivas, con un amplio citoplasma eosinófilo, núcleo excéntrico de cromatina vesicular y nucléolo evidente (Fig 2). Entre ellas se advertía una matriz mixoide, abundantes estructuras vasculares y numerosos polinucleares neutrófilos (Fig 3). Las mitosis eran frecuentes y atípicas. El estudio inmunohistoquímico demostró que las células neoplásicas eran fuertemente positivas para CD68 (Fig 4) y lisozima, y negativas para marcadores linfoides B y T, citoqueratinas, AME, proteina S-100, HMB45, mieloperoxidasa, CD1a y marcadores de células dendríticas (CD21 y CD35). Se hizo el diagnóstico de sarcoma histiocítico (linfoma histiocítico verdadero). No había leucemia, y la biopsia de médula ósea resultó negativa para células neoplásicas.
La paciente recibió radioterapia local (40 Gy en veintidós sesiones), sin respuesta clínica. A los pocos meses aparecieron nuevas adenomegalias (inguinal derecha, retroperitoneales y periesplénicas) y una lesión ocupante de espacio de 3.5 cm de diámetro en la porción posterior de la articulación tibio-astragalina derecha (Fig 5) Se realizó PAAF de la lesión del tobillo (Fig 6) con idéntico resultado a la punción inicial del ganglio linfático. En la tomografía por emisión de positrones (PET) se apreciaron depósitos patológicos del trazador en los tejidos blandos de la raiz del muslo, regiones inguinal e ilíaca derechas y tobillo derecho.
Se inició tratamiento quimioterápico con CHOP (4 ciclos), sin respuesta clínica por lo que se decidió sustituirlo por el protocolo MINE+G (seis ciclos), con el mismo resultado negativo. Dos años después del comienzo de la enfermedad había nuevos focos patológicos en el abdomen y el miembro inferior derecho (Fig 7). Uno de estos últimos fue biopsiado para comprobar que se trataba del mismo sarcoma histiocítico (Fig 8). Se inició entonces una tercera línea de quimioterapia con taxol y topotecan (tres ciclos) y se administró radioterapia paliativa sobre el tobillo. Finalmente, además de las medidas paliativas, se empleó quimioterpia con CVP (tres ciclos) y fludarabina intravenosa. La paciente falleció a los tres años del comienzo de la enfermedad.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
El sarcoma histiocítio (SH), anteriormente conocido como linfoma histiocítico verdadero (1), es una neplasia poco frecuente, que presenta rasgos morfológicos e inmunohistoquímicos de diferenciación histiocítica. Hoy sabemos que la mayor parte de los casos que en el pasado (antes de los últimos quince años) se consideraron como linfomas histiocíticos eran en realidad linfomas B de células grandes o linfomas T anaplásicos. Clínicamente pueden presentarse bien con afectación ganglionar (en un tercio de los casos), cutánea (como lesión solitara o múltiple) o de otros territorios extraganglionares, particularmente el tracto intestinal, pero se han descrito casos en las más diversas localizaciones (tejidos blandos, cavidad nasal, pulmones, testículos...).
Aunque el aspecto cito-histológico de la neoplasia (grandes células epitelioides no cohesivas con abundante citoplasma eosinófilo, núcleo vesicular y nucléolo prominente) debe sugerirlo, el diagnóstico descansa en un estudio inmunohistoquímico que excluya otras neplasias y confirme la naturaleza histiocítica del tumor. El diagnóstico diferencial, conceptualmente más complejo, con las neoplasias de células dendríticas y el sarcoma /histiocitosis de células de Langerhans puede hacerse, según Pileri et al (2), con un panel de tan sólo seis anticuerpos, según se muestra en la tabla siguiente:
Muy recientemente el grupo de hematopatología del AFIP ha comunicado la especificidad de línea histiocítica del nuevo marcador inmuhistoquímico CD163, y su utilidad en el diagnóstico del SH (3).
El SH tiene un curso clínico agresivo y se asocia a una corta supervivencia, generalmente inferior a los dos años desde el momento del diagnóstico. En su tratamiento, además de la cirugía y la radioterapia, se emplean los protocolos quimioterápicos propios de los linfomas agresivos de células grandes, casi siempre con escasa respuesta clínica (4). En nuestro caso se administraron numerosos ciclos de varios de estos protocolos sin que pudiera evitarse la progresión de la enfermedad y el fallecimiento de la paciente.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
1. Copie-Bergman Ch, Wotherspoon AC, Norton AJ, Diss TC, Issacson PG. True histiocytic lymphoma. A morphologic, immunohistochemical, and molecular genetic study of 13 cases. Am J Surg Pathol 1998;22:1386-1392.
2. Pileri SA, Grogan TM, Harris NL, et al. Tumours of histiocytes and accesory dendritic cells: an immunohistochemical approach to classification from the International Lymphoma Study Group based on 61 cases. Histopathology 2002;41:1-29.
3. Vos JA, Abbondanzo SL, Barekman CL, Andriko JW, Miettinen M, Aguilera NS. Histiocytic sarcoma: a study of five cases inclkuding the histiocyte marker CD163. Mod Pathol 2005;18:693-704.
4. Hornick JL, Jaffe ES, Fletcher ChDM. Extranodal histiocytic sarcoma. Clinicopathologic analysis of 14 cases of a rare epithelioid malignancy. Am J Surg Pathol 2004;28:1133-1144.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
- Oscar Marin (09/10/2005 23:35:23)
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
.jpg) fiogf49gjkf0dFig 1. Ganglio linfático. Arquitectura ganglionar parcialmente borrada. HE">
fiogf49gjkf0dFig 1. Ganglio linfático. Arquitectura ganglionar parcialmente borrada. HE">
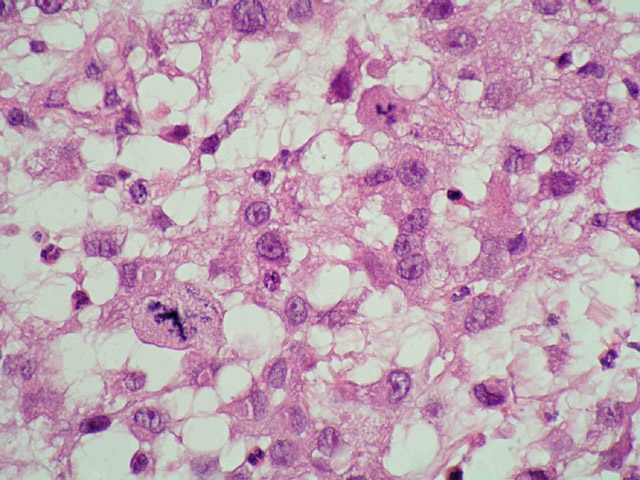 fiogf49gjkf0dFig 2. Ganglio linfático. Celularidad neoplásica. HE">
fiogf49gjkf0dFig 2. Ganglio linfático. Celularidad neoplásica. HE">
.jpg) fiogf49gjkf0dFig 3. Ganglio linfático. Estroma mixoide entre células neoplásicas no cohesivas. HE">
fiogf49gjkf0dFig 3. Ganglio linfático. Estroma mixoide entre células neoplásicas no cohesivas. HE">
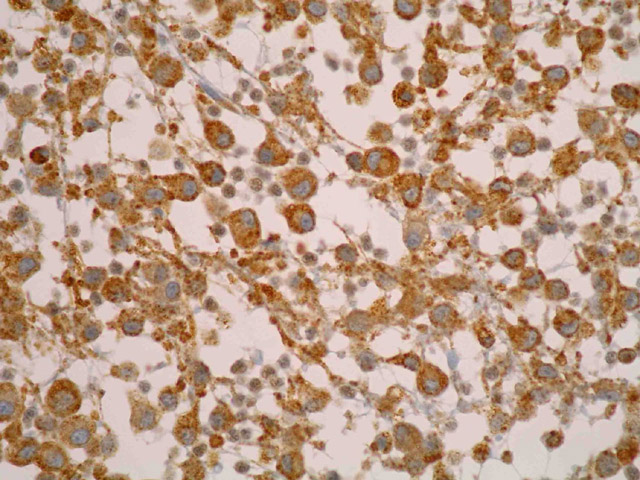 fiogf49gjkf0dFig 4. Ganglio linfático. Células neoplásicas positivas con D68. IHQ">
fiogf49gjkf0dFig 4. Ganglio linfático. Células neoplásicas positivas con D68. IHQ">
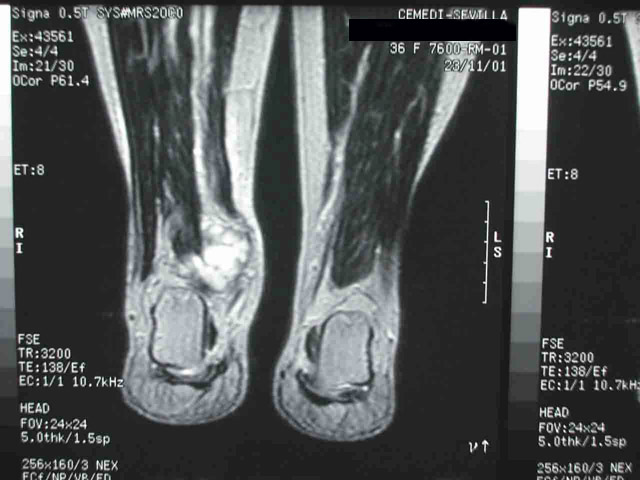 fiogf49gjkf0dFig 5. RNM. Tumoración en los tejidos blandos del tobillo derecho.">
fiogf49gjkf0dFig 5. RNM. Tumoración en los tejidos blandos del tobillo derecho.">
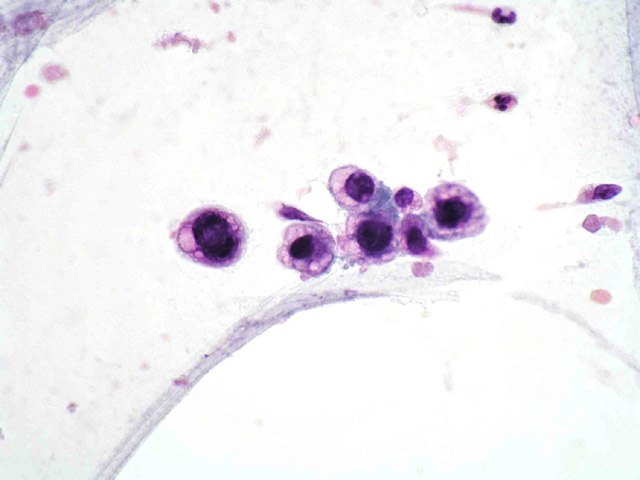 fiogf49gjkf0dFig 6. PAAF de la tumoración del tobillo. Células grandes atípicas, no cohesivas y con vacuolización citoplásmica">
fiogf49gjkf0dFig 6. PAAF de la tumoración del tobillo. Células grandes atípicas, no cohesivas y con vacuolización citoplásmica">
 fiogf49gjkf0dFig 7. Tomografía por emisión de positrones (PET). Depósitos patológicos del trazador en el tobillo derecho.">
fiogf49gjkf0dFig 7. Tomografía por emisión de positrones (PET). Depósitos patológicos del trazador en el tobillo derecho.">
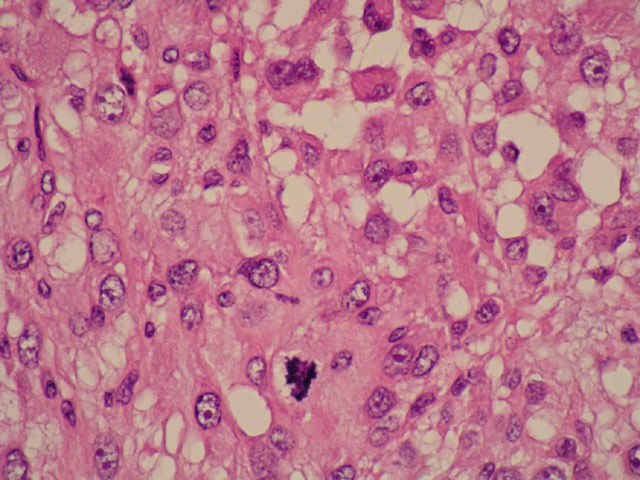 fiogf49gjkf0dFig 8. Tejidos blandos del tobillo. Células neoplásicas iguales a las del ganglio linfático. HE">
fiogf49gjkf0dFig 8. Tejidos blandos del tobillo. Células neoplásicas iguales a las del ganglio linfático. HE">
Web mantenido y actualizado por el Servicio de informática uclm. Modificado: 16/06/2015 15:10:50