Direccion de contacto
fiogf49gjkf0d Julio Merino Montes.
Servicio de Anatomía Patológica.
Hosp. Rafael Méndez Ctra. Nacional, 340 Km 589.
30800 Lorca (Murcia).
jmerinojr@yahoo.es
|
|
Miofibromatosis infantil
Julio Merino Montes*, Cristina Godoy Alba*, Miguel Cámara Pulido*
* Hospital Universitario "San Cecilio". Granada. ESPAÑA
|
|
|
Resumen
fiogf49gjkf0d
Recién nacido varón de 36 semanas de gestación de madre de 30 años secundípara sin antecedentes personales ni familiares de interés, que presenta parada cardiorrespiratoria. Tras maniobras de reanimación, es ingresado en la UCI pediátrica, falleciendo a los 5 días.
El recién nacido presentaba edemas generalizados con múltiples lesiones dérmicas de tipo maculo-papulares de coloración pardo grisácea y consistencia aumentada al tacto. El examen autópsico reveló varias lesiones nodulares en la serosa de intestino delgado y grueso de escasos milímetros de grosor y en estómago, éstas últimas de mayor dimensión.
A microscopio óptico presentan una disposición nodular con un patrón de crecimiento característicamente bifásico constituido en su porción más periférica por células de hábito fusiforme y de citoplasma eosinófilo dispuesto en nódulos, pequeños haces o remolinos. Inmunohistoquímicamente estas zonas son positivas para vimentina y actina pero no para desmina ni para proteína S-100, apoyando por tanto su origen miofibroblástico.
|
|
|
Introduccion
fiogf49gjkf0d La miofibromatosis fue inicialmente descrita por Williams y Schrum en la década de los 50 como fibrosarcoma congénito. Años más tarde Stout la definió como fibromatosis generalizada congénita. Cheng y Enzinger en la década de los 80 renombraron esta enfermedad como miofibromatosis infantil. Actualmente se prefiere usar el término de miofibroma (tres veces más frecuente) o miofibromatosis en función de que las lesiones sean únicas o múltiples. A pesar de ser una entidad rara, constituye el tumor fibroso más frecuente en la infancia.
Las lesiones aisladas presentan muy buen pronóstico, y están constituidas mayoritariamente por nódulos cutáneos que se encuentran habitualmente en la dermis o hipodermis midiendo desde pocos milímetros a varios centímetros, son móviles, presentando una coloracion violácea que puede remedar a un hemangioma. Las lesiones aisladas pueden encontrarse además de la piel en huesos (mandíbula) así como en partes blandas, y más raramente en vísceras.
En pacientes afectos por lesiones múltiples, éstas presentan las mismas características que cuando se manifiestan de forma aislada. Más de un 40% de los pacientes que presentan lesiones viscerales múltiples las tienen ya en el momento del nacimiento. Además de partes blandas y hueso, los lugares más frecuentes de presentación son los pulmones, el corazón, tracto gastrointestinal y páncreas; y raramente el sistema nervioso central, causando la muerte a estos pacientes por diferentes complicaciones a los pocos días o semanas después de su nacimiento. Dichas lesiones crecen en el último periodo de la gestación, continuando el los primeros años de vida.
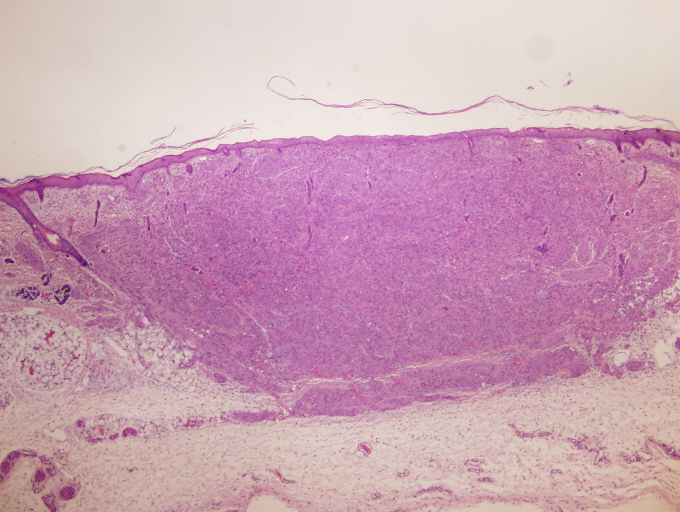
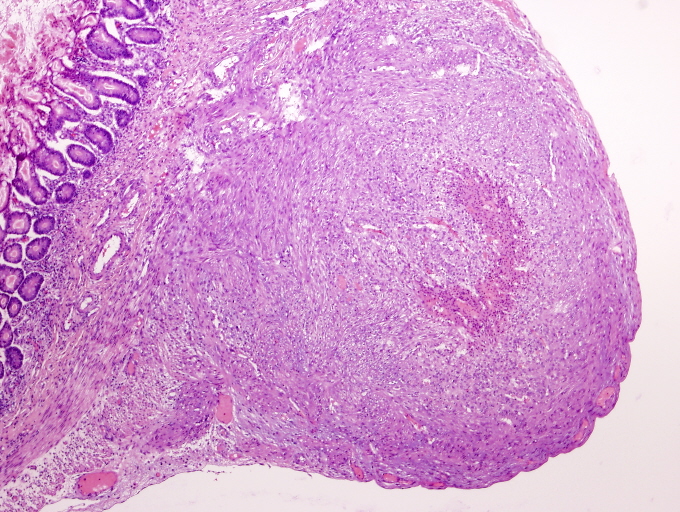
A microscopio óptico presentan una disposición nodular con un patrón de crecimiento característicamente bifásico constituido en su porción más periférica por células de hábito fusiforme y de citoplasma eosinófilo dispuesto en nódulos, pequeños haces o remolinos. Los núcleos muestran una disposición acigarrada sin atipia. Algunas de estas áreas presentan una amplia hialinización. Inmunohistoquímicamente estas zonas son positivas para vimentina y actina pero no para desmina ni para proteína S-100, apoyando por tanto su origen miofibroblástico.
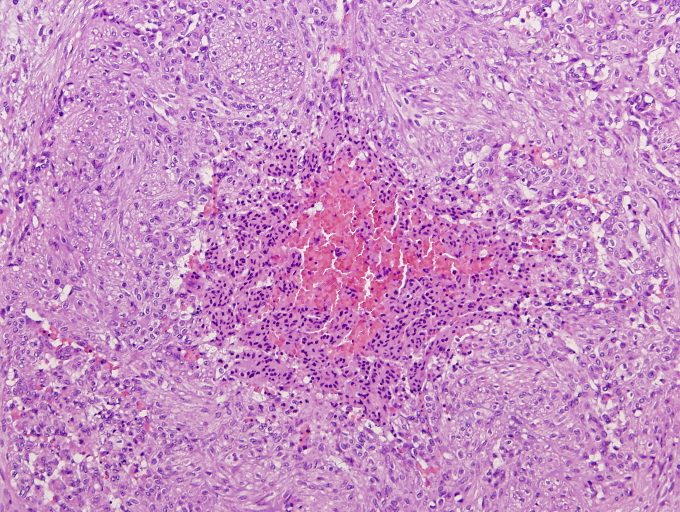
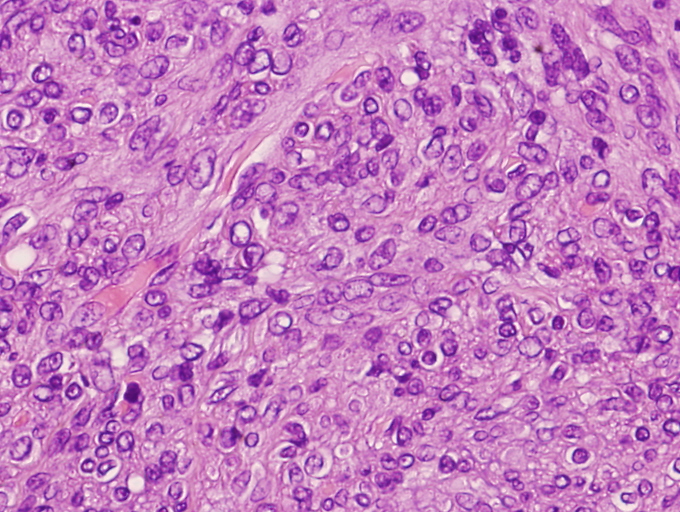
Las áreas centrales están constituidas por células de hábito poligonal o redondeado con núcleos de aspecto más pleomórfico que su contrapartida periférica adoptando un patrón vascular característico tipo hemangiopericitoma. Las células muestran un núcleo vesiculoso con citoplasmas pequeños eosinófilos mal delimitados. Las mitosis son infrecuentes, aunque pueden contabilizarse en torno a 8 mitosis por 10 campos de gran aumento. En algunos casos se pueden observar en estas porciones centrales focos de hemorragia, degeneración quística, o necrosis coagulativa, a menudo acompañado por focos de calcificación. Periféricamente puede observarse un discreto infiltrado inflamatorio tipo crónico.
.jpg) fiogf49gjkf0d"> fiogf49gjkf0d">
- fiogf49gjkf0d
.jpg) fiogf49gjkf0d"> fiogf49gjkf0d">
- fiogf49gjkf0d
|
|
|
Material y Métodos
fiogf49gjkf0d Se realizó estudio necrópsico, procesándose el tejido de forma habitual el cual se estudió ttras tinción de H-E de rutina. El estudio inmunohistoquímico siguió el procedimiento de trabajo usual.
|
|
|
Resultados
fiogf49gjkf0d Recién nacido varón de 36 semanas de gestación de madre de 30 años secundípara sin antecedentes personales ni familiares de interés.
Acude a consulta de su ginecólogo donde se le diagnostica oligoamnios por lo que se deriva para su ingreso a nuestro Hospital.
Tras constatar ecográficamente oligoamnios así como pérdida de bienestar fetal se decide inducir el parto. El recién nacido presenta parada cardiorrespiratoria con una puntuación en el test de APGAR de 0 en el momento del nacimiento y a los 5 minutos y de 3 a los 10 minutos tras maniobras de reanimación, es ingresado en la UCI pediátrica, falleciendo a los 5 días.
El recién nacido presentaba edemas generalizados con múltiples lesiones dérmicas de tipo maculo-papulares de coloración pardo grisácea y consistencia aumentada al tacto. El examen autópsico reveló varias lesiones nodulares en la serosa de intestino delgado y grueso de escasos milímetros de grosor y en estómago, éstas últimas de mayor dimensión.
El examen histológico mostró múltiples lesiones, bien delimitadas, todas de morfología similar, que además de las reseñadas en piel y tracto digestivo se continuaban en pulmones, corazón, hígado, suprarrenales, timo, ganglio linfático y paraganglios. Estas tumoraciones estaban constituidas por haces fusocelulares dispuestos de forma circular o en pequeños remolinos por células de escaso citoplasma eosinófilo mal delimitado y con núcleos acigarrados sin mitosis evidentes. Destacaba un componente central de tipo hemangiopericítico de extensión variable en función del tamaño de la tumoración.
El perfil inmunohistoquímico mostró positividad para vimentina, y actina especifica de músculo liso (ésta última en las áreas periféricas de morfología fusocelular); siendo negativo para pancitoqueratinas, proteína S-100 y EMA.
 fiogf49gjkf0d"> fiogf49gjkf0d">
- fiogf49gjkf0d
 fiogf49gjkf0d"> fiogf49gjkf0d">
- fiogf49gjkf0d
 fiogf49gjkf0d"> fiogf49gjkf0d">
- fiogf49gjkf0d
 fiogf49gjkf0d"> fiogf49gjkf0d">
- fiogf49gjkf0d
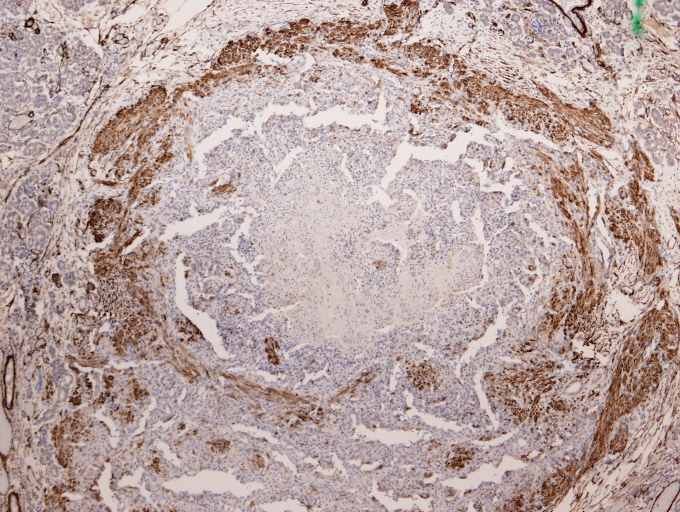 fiogf49gjkf0dActina"> fiogf49gjkf0dActina">
Actina - fiogf49gjkf0d Actina
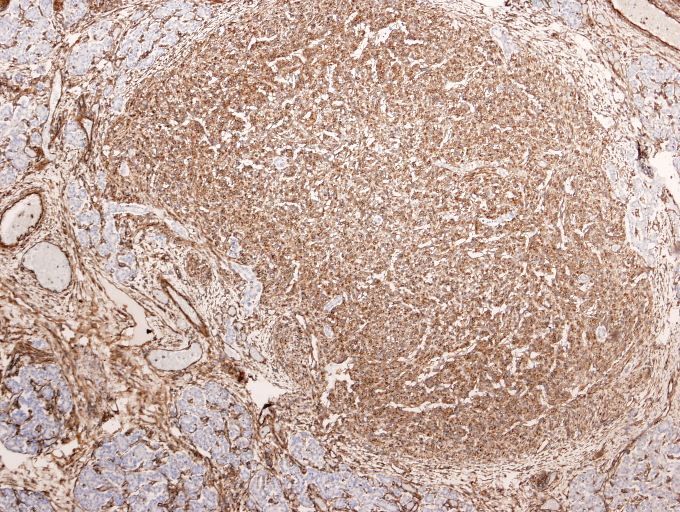 fiogf49gjkf0dDesmina"> fiogf49gjkf0dDesmina">
Desmina - fiogf49gjkf0d Desmina
|
|
|
Discusión y conclusiones.
fiogf49gjkf0d El curso clínico está fuertemente determinado por la extensión de la enfermedad, siendo excelente en los casos solitarios (miofibroma), y de peor pronóstico en los múltiples, estando directamente relacionados con la extensión de las tumoraciones y según los órganos afectados, regresando espontáneamente en los casos que no comprometen la vida del paciente por mecanismos de apoptosis masiva como postuló Fukasawa.
Aunque la miofibromatosis es la tumoración fibrosa más frecuente de la infancia, muchos aspectos de esta lesión siguen aun sin ser completamente comprendidos en la actualidad. A pesar de que los resultados inmunohistoquímicos y ultraestructurales apoyen el origen a partir del miofibroblasto quedan detalles desde el punto de vista histogenético aun por aclarar. La información genética actual es escasa con muy pocos casos cariotipados, estando actualmente aceptado el patrón de herencia autosómica dominante, como mecanismo de transmisión en los casos familiares.
Hasta la fecha se han comunicado dos alteraciones citogenética, si bien ambas en casos de miofibromas solitarios, en la primera se describió una defección de cromosoma 6 (q12; q15), y la segunda en una translocación disbalanceada del brazo completo del cromosoma 9 al 16, resultando una monosomía del cromosoma 9 y trisomía 16.
|
|
|
Bibliografía
-Chung EB, Enzinger FM. Infantile myofibromatosis. Cancer 1981; 48: 1807-1818.
-Weiss SW. Fibrous tumors of infante and childhood. En:Weiss SW. Goldblum JR (Eds) Enzinger and Weiss´s Soft Tissue tumors.St Louis: Mosby, 2001: 357-363.
-Bélenger A, Aparicio JM, Cuevas J, Muñoz A. Miofibromatosis infantil sistemica en un lactante. An Esp Pediatr 2001; 55: 83-86.
-Zand D, UHF D, Everman D, russell K, Saitta S, McDonald-McGinn D, Zacki E. Autosomal dominant inheritance of infantile myofibromatosis. Am J Med Gen 2004; 126A: 261-266.
|
|
|
Comentarios
|
|
|
|
|

