

|
Síndrome mileodisplásico primario. Análisis de las características morfológicas y evolución. Dr. Alejandro S. Ruiz Méndez*, Dr. Daniel Artiles Artiles**, Dra. Lissete Ruiz Jorge**, Dr. Victor Hugo Cortés Rodríguez**, Dr. Manuel Arce González**, Dra Olga Lidia Alonso Mariño***, Dr. José Luis Aparicio Suárez**, Téc. Marlenes Ruiz López** |
|
|
En el presente trabajo se estudió un grupo de 65 pacientes diagnosticados como Síndrome Mielodisplásico y a los cuales se les realizó biopsia de médula ósea en el período comprendido entre Agosto de 1994 y Mayo del 2005, en el Hospital “Arnaldo Milián Castro” de Santa Clara, con el objetivo de conocer las características clínicas y morfológicas del síndrome en nuestro medio, así como resaltar la importancia de los hallazgos de la biopsia de médula ósea en el diagnóstico y evolución de estos pacientes. Entre los resultados obtenidos se encontró que el Síndrome Mielodisplásico predominó en el grupo de pacientes por encima de 60 años con discreto predominio del sexo masculino, la forma de presentación clínica fue mediante la presencia de una o varias citopenias, existió relación entre el mayor número de citopenias y corta sobrevida. Prevaleció el grupo de la AR de la clasificación FAB con breve supervivencia para los grupos AREB y AREBt. Mediante el estudio de la biopsia de médula ósea se ratificó que la hipocelularidad, el fenómeno LAPI y la esclerosis son los hallazgos de la BMO que tienen una relación más estrecha con una corta sobrevida de los enfermos. Las causas de muerte que predominaron fueron la transformación a LMA y la sépsis.
|
||
|
|
El Síndrome Mielodisplásico (SMD) descrito por primera vez en 1953, es una hemopatía de carácter monoclonal que afecta a la célula madre progenitora pluripotencial, con displasia de una o más de las tres líneas de estirpe mieloide y caracterizado por un curso insidioso e inefectividad de la hematopoyesis lo cual ocasiona una o varias citopenias, y tendencia a evolucionar a Leucemia Mieloblástica Aguda. Muestra un ligero predominio del sexo masculino y una incidencia de 20 a 50 casos nuevos por cada 100 000 hab. mayores de 60 años. La sobrevida media después de realizado el diagnóstico es de 24 meses. (1, 2, 3, 4, 5, 6)
Se diferencian fundamentalmente dos grupos de SMD, uno primario, idiopático o de novo y otro menos frecuente secundario a la exposición de agentes mutágenos, siendo los más frecuentes los productos de la quimioterapia, las radiaciones y los compuestos órgano fosforados de la agricultura. (4, 7, 8)
El diagnóstico del SMD es por exclusión y se basa en: la anamnesis, el examen físico, el medulograma, la biopsia de médula ósea y estudios citogenéticos; la anamnesis, el exámen físico y la morfología nos permiten hacer un diagnóstico definitivo en la mayoría de los casos de la enfermedad, en tanto que los estudios citogenéticos, aunque muestran una gran especificidad, su alto costo no permite la generalización, mucho más aun, cuando el grupo etáreo en que se presenta este síndrome, tiene una alta incidencia de otras enfermedades que cursan con anemia. (5, 7, 9, 10, 11, 12, 13, 15)
Esta situación ha estimulado el uso de la biopsia de médula ósea para establecer el diagnóstico y emitir un pronóstico del SMD, cuya efectividad es superior al 90%. (3, 8, 15)
Entre las principales alteraciones que aparecen en la médula ósea y que permite el diagnóstico de SMD se encuentran: la pérdida de la arquitectura, la Localización Anormal de Precursores Inmaduros conocido como fenómeno LAPI, la existencia de una trama de reticulína, demostrada por técnica de retículo, distribución paratrabecular de la serie eritroide y megacariopoyética y alteraciones del estroma menos específicas como la presencia de células plasmáticas, eosinófilos, linfocitos y mastocitos, degeneración serosa del estroma, dilatación de capilares y acumulación de hierro intra o extra celular. (8, 6, 17)
A pesar de la utilidad que ha demostrado la biopsia de médula ósea en el diagnóstico y pronóstico de esta enfermedad, todas las clasificaciones realizadas hasta la fecha (FAB, OMS, IPSS, etc.) obvian los hallazgos morfológicos de la biopsia, dándole solo importancia a los datos aportados por el medulograma, los estudios citogenéticos y variables clínicas como el número de citopenias. (18,19, 20)
El objetivo de nuestro estudio consiste en caracterizar el comportamiento clínico del SMD de nuestros casos, su distribución según la clasificación FAB y demostrar la relación existente entre los hallazgos morfológicos en la médula ósea y la sobrevida alcanzada.
|
|
|
|
El presente trabajo es un estudio retrospectivo, efectuado en el Hospital “Arnaldo Milián Castro” de la ciudad de Santa Clara, e incluyó a todos aquellos pacientes con diagnóstico de Síndrome Mielodisplásico primario (SMDp) a los cuales se les realizó Biopsia de Médula Ósea, en el período comprendido entre Agosto de 1994 y Mayo del 2005.
Para la selección de la muestra y recolección de los datos se utilizaron, los resultados obtenidos en las biopsias de médula ósea efectuada en ese período y archivadas en el departamento de Anatomía Patológica. Luego se revisó la historia clínica procedente del archivo central hospitalario y los informes del medulograma. Se realizaron sesiones de entrevistas con los hematólogos que atendieron a los pacientes y por último se procedió a contactar directamente con los enfermos o sus familiares, para conocer el estado actual de los mismos, y en caso de haber fallecido conocer la fecha, la causa y si se le realizó estudio necrópsico.
Fue motivo de exclusión, la no realización de BMO, también se excluyeron 2 pacientes que no pudieron ser contactados para conocer su evolución.
Después de cumplido los requisitos, la muestra quedó conformada por un universo de 65 pacientes, que se subdividieron en dos grupos, uno de 43 pacientes fallecidos y el segundo por 22 pacientes vivos en el momento del estudio. Para el análisis de las variables estudiadas se excluyeron también a 4 pacientes con el diagnóstico de Leucemia Mielomonocítica Crónica, esta decisión se tomó debido a que la inclusión de esta entidad dentro de la clasificación FAB siempre fue motivo de controversias por sus características clínico/hematológicas y en lo que respecta a nuestro trabajo, no permitía evaluar las variables fundamentales en que se apoya la biopsia de médula ósea, o sea: la celularidad, el fenómeno LAPI y la esclerosis.
A todos se les realizó una encuesta para recoger las variables a estudiar.
La encuesta se dividió en 8 secciones fundamentales:
1. Datos generales.
2. Anamnesis y Examen físico.
3. Valores hematológicos de laboratorio.
4. Resultado de los medulogramas.
5. Evolución con el tiempo de transformación.
6. Hallazgos de la Biopsia de Médula Ósea.
7. Estudio de la autopsia.
8. Causas de la muerte.
Los datos generales, la anamnesis y el examen físico, se obtuvieron de las historias clínicas hospitalarias. Para la edad del diagnóstico del SMDp, se tomó como referencia la edad en que se realizó el diagnóstico ya sea por medulograma o por BMO. En caso de no haber fallecido se tomó como fecha de última consulta la realizada en el año 2005 o en su defecto la comunicación directa con el paciente o los familiares que permitió saber el estado actual del enfermo.
En los complementarios hematológicos, se consideró anemia los valores de Hb, por debajo de 12.0 g/l, leucopenia por debajo de 5 000 células x mm³ y leucocitosis por encima de 10 000 células x mm³. Para la evaluación del número de plaquetas se utilizó el rango de 150 000 plaquetas x mm³ a 300 000 plaquetas x mm³, se señaló como trombocitopenia el valor por debajo de 150 000 y trombocitosis por encima de 300 000.
Se definió como bicitopenia cuando se presentaba la anemia acompañada por leucopenia o trombocitopenia y tricitopenia cuando se encontraban las tres series disminuidas en un mismo paciente.
Las causas de muerte fueron divididas en relacionadas o no con la enfermedad, en el primer caso se incluyen la sépsis, los sangramientos, y la descompensación cardiovascular secundaria a la anemia.
El estudio de la biopsia de médula ósea se realizó sobre cortes de cilindros de médula ósea incluidos en parafina, a los cuales se les realizaron tres técnicas histoquímicas: Hematoxilina/Eosina, técnica especial de Pers para demostración de gránulos de hierro y tricrómica de Retículo de Gomori para evaluar el nivel de organización del retículo. Los datos obtenidos por la BMO fueron la celularidad, el fenómeno LAPI, la esclerosis y las alteraciones estromales.
Para determinar la celularidad, se tuvo en consideración, la variabilidad que muestra la relación entre el tejido hematopoyético y el tejido graso en las distintas etapas de la vida, la cual ocsila, desde un 85% de tejido hematopoyético en la infancia, hasta un 30% en la vejez, con una aproximación del 50% de ambos tejidos en la edad adulta.
Se planteó hipocelularidad cuando el tejido hematopoyético era inferior a un 15% de lo estimado para la edad y se consideró hipercelular cuando aumentó un 15% a lo esperado según la edad.
LAPI: es la presencia de grupos de mieloblastos y promielocitos en número de 5 a 8 células en la porción central del espacio medular, alejado de estructuras vasculares o superficies endostiales de la trabécula ósea. LAPI menor es cuando se identifican hasta dos grupos de estas células en un espacio medular. LAPI mayor cuando se identifican más de dos grupos en algún espacio medular.
La esclerosis medular se determina con la utilización de la técnica de Retículo de Gomori, en estado normal solo se observa a nivel de la pared de los vasos sanguíneos, este hecho es normal y por tanto se considera como retículo adecuado. Cuando esta se distribuye de forma radiada pero limitada a la periferia de los vasos sanguíneos se denominó como esclerosis temprana y el término de esclerosis avanzada se refiere a la formación de una red reticulínica generalizada con finas prolongaciones que se entretejen entre las células.
Para exponer los resultados se utilizan tablas y gráficos con análisis porcentual.
|
|
|
|
Análisis y Discusión de los Resultados En el período analizado se hicieron un total de 970 biopsias de médula ósea de las cuales se diagnosticaron como Síndrome Mielodisplásico primarios un total de 65 pacientes para un 6.7%. La distribución por grupo de edades se representa en la (Tabla 1) donde vemos que el 72.3% son enfermos mayores de 60 años de edad, este por ciento es ligeramente inferior al reportado en otros trabajos en los que alcanzan hasta el 80%, lo que pueden estar relacionado con que algunas de las anemias en los ancianos no se estudian con la profundidad requerida. El 21,5% correspondió a las edades de 40 a 60 años y un 6.2% a los menores de 40 años de edad. Esto demuestra que el SMD predomina en los pacientes ancianos donde al mismo tiempo es la hemopatía maligna más frecuente. La edad media fue de 66.07 años. Y los extremos de edades observados fueron un niño de 8 años y una anciana de 93 años; existen reportes en la literatura incluso con edades inferiores. En esta misma tabla se representa la división de los pacientes según el sexo, los cuales muestran una relación de 1.4 hombre por cada mujer. Aunque para la mayoría de los autores el sexo no tiene una influencia reconocida en la frecuencia de la enfermedad, todos coinciden con una prevalencia ligeramente superior para el sexo masculino que llega a ser de hasta 2.6 por cada mujer en algunos reportes. La distribución según la clasificación FAB está expresada en la (Tabla 2), La AR se encontró en el 67.7%; seguida por la AREB con 10.8%, luego la AREBt con 12.3%, la ARS solo se observó en 2 pacientes lo que representó un 3.1%. La LMMC se encontró en 4 casos para un 6.2%. De los 65 casos con SMD fallecieron 43 (60%) y se encuentran vivos 22 (40%). A partir de este momento se analizan diferentes variables en los grupos AR, ARS, AREB y AREBt y se excluye la LMMC (4 pacientes), debemos recordar que la inclusión de esta última categoría en los SMD por las FAB ha sido motivo de críticas por tener características distintivas que no se ajustan al resto de los SMD, de hecho la OMS creó una categoría aparte para esta entidad. Por todo ello el número total que se analiza es de 61 (22 vivos y 39 fallecidos). En la (Tabla 3) se distribuye el número de citopenias entre los pacientes vivos y fallecidos y puede observarse que la presencia de 2 o 3 citopenias se ubican en su mayoría en el grupo de los fallecidos con 83.3% y 78.9% con una diferencia altamente significativa en relación a los vivos. La presencia de una citopenia no representa una diferencia significativa entre ambos grupos. En la (tabla 4) se relaciona el número de citopenias con el tiempo de sobrevida de los fallecidos, la presencia de 2 y 3 citopenia se relacionó con una corta sobrevida al encontrarse el 70% y el 66.7% fallecidos antes de los 12 meses de trascurridos desde el diagnóstico. Si sumamos este grupo con el que vivió de 12 a 24 meses entonces se alcanza la cifra del 80% para ambos grupos, lo cual reafirma el mal pronóstico que significa tener más de una citopenia. En la (Tabla 5) se distribuyen por el número de citopenias de los fallecidos según la clasificación FAB y vemos que el 60% de las tricitopenias se agrupan entre las anemias refractarias que constituyen junto a las anemias refractarias con sideroblastos los grupos de mejor pronóstico de esta clasificación, en el caso de la clasificación FAB no existe una distinción entre aquellos pacientes con una anemia refractaria aislada y aquellos con más de una citopenia. En la (Tabla 6) se distribuyen los casos según el grado de celularidad encontrado en la BMO en los grupos de pacientes vivos y fallecidos; observamos que la mayor frecuencia estuvo en el grupo de las médulas óseas hipercelulares con el 68.9%, lo que es un hallazgo común en este trastorno, seguido de las normocelulares con 19.7% y por último las hipocelulares con 11.5%. La hipocelularidad se describe en un rango del 10 al 20% y constituyen los casos mas difíciles de diagnosticar por el medulograma al acompañarse de aspirados insuficientes, mientras en la BMO es necesario diferenciarlo de los estados de insuficiencia medular además de ser mas difícil apreciar el fenómeno LAPI. En nuestros casos la hipocelularidad se ubicó en su mayoría en el grupo de fallecidos (85.7%). La normocelularidad se distribuyó de manera uniforme entre ambos grupos con discreto predominio en el grupo de los pacientes vivos, alcanzando un 58.3% en relación al 41.7% en el grupo fallecido. La (tabla 7) relaciona la celularidad y el tiempo de sobrevida en fallecidos y se observa que la mayoría de los pacientes con hipercelularidad (67.9%) e hipocelularidad (83.3%) fallecieron en los primeros 12 meses después del diagnóstico, destacándose que todos los hipocelulares fallecieron antes de los 24 meses. En sentido contrario, tenemos que todos los pacientes con normocelularidad fallecieron después de los 12 meses de realizado el diagnóstico. En la (tabla 8) se analiza la distribución del fenómeno LAPI en vivos y fallecidos. De los 61 casos de la serie en 20 (32.8%) no se encontró el fenómeno. En 41 (67.3%) se pudo apreciar LAPI; de los cuales en 14 (23%) lo fue en grado menor y en 27 (44.3%) en grado mayor. Como puede observarse de los que mostraron el fenómeno LAPI en grado mayor fallecieron 25 para un 92.6% en comparación con 2 (7.4%) que están vivos, lo que representa una diferencia notable. Los casos con LAPI menor se distribuyeron de forma uniforme en ambos grupos. La ausencia de LAPI se distribuyó con mayor número de casos entre los pacientes vivos 13 (65%). En la (tabla 9) se distribuye el fenómeno LAPI según el grado y el tiempo de sobrevida. Se puede observar que de los pacientes con fenómeno LAPI mayor, 17 (68%) fallecieron en el primer año posterior al diagnóstico, lo cual contrasta con el 8% y 24% que sobrevivieron entre 12 y 24 meses o más de 24 meses respectivamente lo que demuestra una relación directa entre la magnitud del fenómeno LAPI y la corta sobrevida. Por otra parte 19 casos (76%) fallecen en los primeros dos años después del diagnóstico. En este punto hay que hacer mención sobre el hecho que de los tres pacientes sin fenómeno LAPI y que fallecieron antes de los 12 meses dos correspondían a la variedad hipocelular, al igual que en el caso del LAPI menor, dos de los cuatro pacientes mostraron hipocelularidad. La presencia de hipocelularidad disminuye la presencia del fenómeno LAPI, sin embargo, la presencia de hipocelularidad es en sí misma un hallazgo de mal pronóstico. En la (tabla 10) se representa la distribución del fenómeno LAPI en relación a la clasificación FAB, podemos observar como el 100% de los pacientes con ausencia de LAPI clasificaron como AR. Llama la atención, que el 57.1% del LAPI menor y el 44.0% del LAPI mayor se ubicaron en el grupo de AR, lo cual no concuerda con la verdadera gravedad de su hallazgo en la BMO. Por otra parte todos los pacientes que estuvieron, en el grupo AREB o AREBt de la clasificación FAB mostraron la presencia del fenómeno LAPI en alguna magnitud, y la mayoría de ellos en grado mayor. En la (tabla 11), se estudió la presencia de esclerosis detectada mediante la técnica para retículo en el grupo de pacientes vivos y fallecidos. Al igual que en la tabla que analiza la relación entre fallecidos y vivos con el fenómeno LAPI, en esta se observa como el 94.4% de los pacientes con esclerosis avanzada se encuentran fallecidos en el momento del estudio, solo se encontraron vivos el 5.6%, de forma parecida, la esclerosis temprana se asoció con un 63.2% de mortalidad y 36.8% entre los pacientes vivos. La ausencia de esclerosis tuvo un comportamiento similar en ambos grupos. La (tabla 12) señala la relación existente entre la presencia de esclerosis y la sobrevida medida en meses posterior al diagnóstico, se observa como la presencia de esclerosis avanzada se asocia a una temprana mortalidad para un 78.9%, en tanto que solo un 15.8% sobrevive más de 24 meses. La ausencia de esclerosis y la esclerosis temprana tuvieron un comportamiento similar. En la (tabla 13) se puede observar la relación entre la edad y causas de muerte. En nuestro estudio la transformación a LMA resultó la causa de muerte más común con 23 pacientes para un 58% del total de fallecidos, con una mayor presencia por encima de 70 años. Le sigue en orden de frecuencia la sépsis a distintos niveles sin presentar una diferencia notable en los distintos grupos de edades.
|
|
|
|
1. El síndrome es más común por encima de los 60 años, con un discreto predominio del sexo masculino.
2. La presencia de más de una citopenia se relaciona con una temprana mortalidad.
3. La hipocelularidad medular es un hallazgo que se asocia a una elevada mortalidad seguido por la hipercelularidad.
4. La presencia del fenómeno LAPI se relaciona con corta sobrevida, en relación directa con la magnitud del fenómeno.
5. El grado de esclerosis avanzada constituye un hallazgo relacionado con corta sobrevida.
6. La causa de muerte más frecuente resultó la Leucemia Mieloide Aguda.
|
|
|
|
1. Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR. Proposals, for the classification of the myelodisplastic syndromes. Br J Haematol 1982; 51:189-199.
2. Jaffe ES, Harris NL, Vardiman JW. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Haematopoietic and lymphoid tissues. Lyon: IARC Press; 2001.
3. Mirza I, Garzon R, Burns J, Edwards L, Fernandez-Cymering C, Kloss R. Detection of risk groups in myelodysplastic syndromes. Haematologica 2002; 87:9-16.
4. Aster J, Kumar V. Síndromes Mielodisplásicos. En: Robbins Patología estuctural y funcional. Madrid: Mc Graw-Hill. Interamericana 2000.p 709-711.
5. Alessandrino EP, Amadori S, Cazzola M. Myelodysplastic syndromes: recent advances. Haematologica 2001; 86:1124-1157.
6. Zhao WL, Xu L, Wu W, Yan H, Tang W, Hu J, et al. The myelodysplastic syndromes: analysis of prognostic factors and comparison of prognostic systems in 128 Chinese patients from a single institution. Hematol J 2002; 3(3):137-44.
7. Olney HJ, Le Beau MM. The cytogenetics and molecular biology of the myelodysplastic syndromes. In: Bennett JM (ed). The Myelodysplastic Syndromes, Pathobiology and Clinical Management. Marcel Dekker, Inc: New York; 2002.
8. Bowen D, Culligan D, Jowitt S. Guidelines for the diagnosis and therapy of adult myelodysplastic syndromes. Br J Haematol. 2003;120:187–200.
9. Arber DA, Stein AS, Carter NH. Prognostic impact of acute myeloid leukemia classification. Importance of detection of recurring cytogenetic abnormalities and multilineage dysplasia on survival. Am J Clin Pathol 2003;119:672–680.
10. Cermak J, Michalova K, Brezinova J. A prognostic impact of separation of refractory cytopenia with multilineage dysplasia and 5q- syndrome from refractory anemia in primary myelodysplastic syndrome. Leuk Res 2003;27:221–229.
11. Greenberg P, Cox C, Le Beau MM. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997;89:2079–2088.
12. Pellagatti A, Esoof N, Watkins F. Gene expression profiling in the myelodysplastic syndromes using cDNA microarray technology. Br J Haematol 2004;125:576–583.
13. Benetatos L, Bourantas KL. Myelodysplastic síndromes. Haema 2005; 8: 21-36.
14. Sole F, Espinet B, Sanz GF, Cervera J, Calasanz MJ, Luno E, et al. Incidence, characterization and prognostic significance of chromosomal abnormalities in 640 patients with primary myelodysplastic syndromes. Haematol 2000 Feb;108(2):346-56
15. Brunning RD, Matutes E, Harris NL. Acute myeloid leukemia: introduction. In: Jaffe ES, Harris NL, Stein H, Vardiman JW. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of the Haematopoietic and Lymphoid Tissues. IARC Press: Lyon; 2001:77–105.
16. Bain BJ. The bone marrow aspirate of healthy subjects. Br J Haematol 1996;94:206–209.
17. Sperr WR, Wimazal F, Kundi M, Fonatsch C, Thalhammer-Scherrer R, Schernthaner GH, et al. Survival analysis and AML development in patients with de novo myelodysplastic syndromes: comparison of six different prognostic scoring systems. Hematol 2001 May;80(5):272-7.
18. Belli C, Acevedo S, Bengio R, Arrossagaray G, Watman N, Rossi N, et al. Detection of risk groups in myelodysplastic syndromes. A multicenter study. Haematologica 2002 Jan;87(1):9-16.
19. Jaiyesimi I, Friedline J, Mattson J, Gyorfi T, Davis B, Al-Khalili A, et al. Myelodysplastic syndrome at a large tertiary care community hospital: analysis according to the international prognostic scoring system. Leuk Res 2000 May;24(5):417-26.
20. Stetler-Stevenson M, Arthur DC, Jabbour N. Diagnostic utility of flow cytometric immunophenotyping in myelodysplastic syndromes. Blood 2001;98:979–987.
|
|
|
|
- Oscar Marin (09/10/2005 23:18:17)
|
|
|
|
|
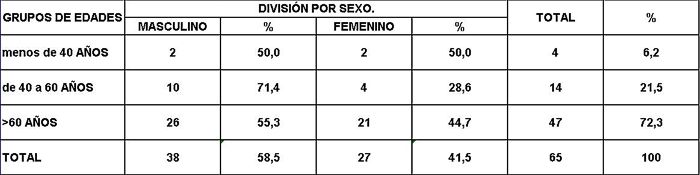 fiogf49gjkf0dRelación entre sexo y edad.">
fiogf49gjkf0dRelación entre sexo y edad.">
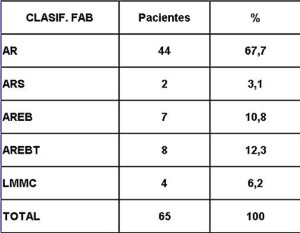 fiogf49gjkf0dDistribución de los pacientes según la clasificación FAB.">
fiogf49gjkf0dDistribución de los pacientes según la clasificación FAB.">
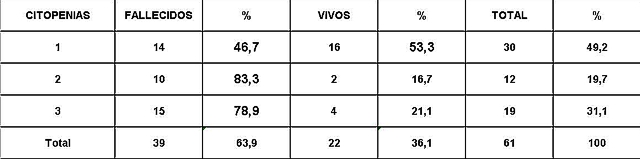 fiogf49gjkf0dNúmero de citopenias entre los pacientes vivos y fallecidos.">
fiogf49gjkf0dNúmero de citopenias entre los pacientes vivos y fallecidos.">
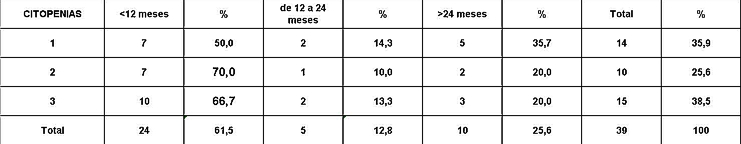 fiogf49gjkf0dRelación entre número de citopenias y sobrevida en meses.">
fiogf49gjkf0dRelación entre número de citopenias y sobrevida en meses.">
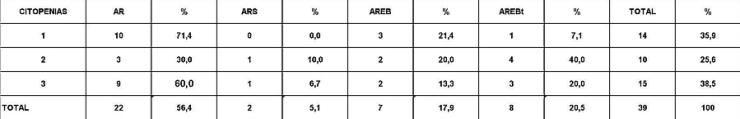 fiogf49gjkf0dRelación entre número de citopenias y clasificación FAB.
">
fiogf49gjkf0dRelación entre número de citopenias y clasificación FAB.
">
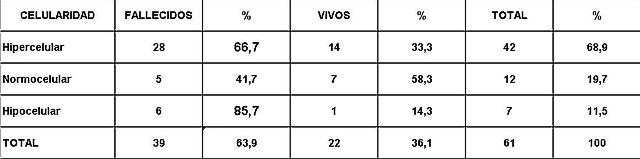 fiogf49gjkf0dGrado de celularidad entre pacientes vivos y fallecidos.">
fiogf49gjkf0dGrado de celularidad entre pacientes vivos y fallecidos.">
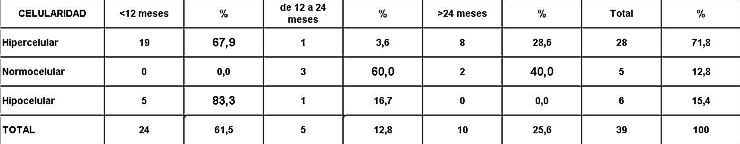 fiogf49gjkf0dRelación entre celularidad y sobrevida en meses.">
fiogf49gjkf0dRelación entre celularidad y sobrevida en meses.">
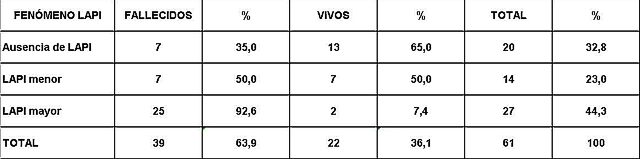 fiogf49gjkf0dPresencia del fenómeno LAPI entre pacientes vivos y fallecidos.">
fiogf49gjkf0dPresencia del fenómeno LAPI entre pacientes vivos y fallecidos.">
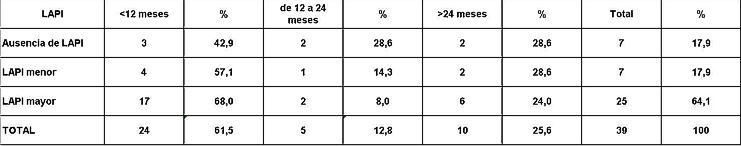 fiogf49gjkf0dRelación entre el fenómeno LAPI y sobrevida en meses.">
fiogf49gjkf0dRelación entre el fenómeno LAPI y sobrevida en meses.">
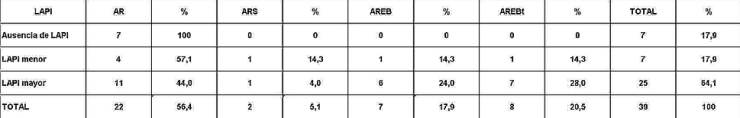 fiogf49gjkf0dRelación entre fenómeno LAPI y clasificación FAB.
">
fiogf49gjkf0dRelación entre fenómeno LAPI y clasificación FAB.
">
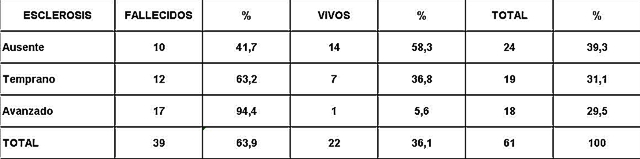 fiogf49gjkf0dRelación entre grado de esclerosis en pacientes vivos y fallecidos.">
fiogf49gjkf0dRelación entre grado de esclerosis en pacientes vivos y fallecidos.">
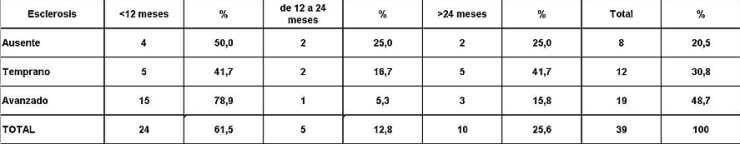 fiogf49gjkf0dRelación entre grado de esclerosis y sobrevida en meses.">
fiogf49gjkf0dRelación entre grado de esclerosis y sobrevida en meses.">
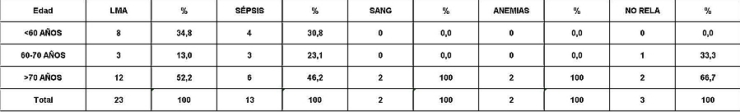 fiogf49gjkf0dRelación entre edad y causa de muerte.
">
fiogf49gjkf0dRelación entre edad y causa de muerte.
">
Web mantenido y actualizado por el Servicio de informática uclm. Modificado: 16/06/2015 15:10:50