
 Comunciación libre
Comunciación libre
|
POLIPO DE PEUTZ-JEGHERS COMO CAUSA DE ABDOMEN AGUDO. DETALLES CLINICOS Y MORFOLOGICOS. Daniel Jesús Piccinni*, Javier Fuentes**, Pablo Fernandez Mancuzo**, Luis Santos Spitale***, María Bachureck** |
|
El síndrome de Peutz jeghers (SPJ) es una rara afección familiar con transmisión autosómica dominante, caracterizada por poliposis hamartomatosa digestiva y lentiginosis cutánea-mucosa con predominio periorificial. Está asociado a una alta incidencia de recurrencia y con un riesgo significativo de malignidades. También, es responsable de complicaciones intestinales como la invaginación y hemorragia, con la que debutan clínicamente la mayoría de los pacientes.Los pólipos tipo Peutz-Jeghers solitarios, con historia familiar negativa y sin hiperpigmentation muco-cutánea, son una entidad rara que normalmente tiene las complicaciones intestinales del SPJ, pero no el riesgo de cáncer gastrointestinal.Reportamos el caso de una mujer 19 años de edad, con manchas lentiginoas en la cara a predominio periorificial (boca y nariz), que debutó clínicamente con un abdomen agudo. En la laparotomía exploradora se detectó una invaginación ileo-ileal con compromiso vascular del segmento invaginado. Al realizar la enterotomía se visualizó un pólipo de 4 cm de diámetro, que fue resecado mediante una enterectomía segmentaria.En el laboratorio de patología se estudió un segmento intestino de 70 cm de largo, que incluía un pólipo rojo-vinoso de 3.8 cm de diámetro.Las secciones histológicas mostraron un pólipo con glándulas hamartomatosas, separadas en grupos por fibras musculares lisas. El pólipo y una porción del intestino resecado presentaban infarto hemorrágico.El diagnóstico patológico final fue de pólipo hamartomatoso tipo Peutz-Jeghers.Creemos que nuestro caso pertenece al SPJ, debido a la acentuada lentiginosis cutánea facial con predominio periorificial que mostraba la paciente, característica que no se ve en el pólipo tipo Peutz-Jeghers solitario.
|
||
|
|
El síndrome de Peutz-Jeghers fue primeramente descrito por Peutz en 1921, como pólipos gastrointestinales y pigmentación mucocutánea, en 7 casos de una misma familia, ocurridos en tres generaciones. Fue confirmado y publicado por Jeghers en 1949, quien reportó la revisión de 10 casos, por lo cual hoy se reconoce y acepta la combinación de los nombres en el síndrome de Peutz-Jeghers (1, 2). Es una rara afección familiar con transmisión autosómica dominante, caracterizada por poliposis hamartomatosa digestiva y lentiginosis muco-cutánea con predominio periorificial. Representa del 3 al 10% de los pólipos digestivos familiares, siendo éstos la parte más importante del SPJ, ya que ellos determinan la manifestación clínica y el pronóstico de la enfermedad, al ser responsables de complicaciones como la invaginación y hemorragia intestinal (3-7). Además, pólipos muy grandes pueden causar obstrucción intestinal mecánica (8).
El SPJ está asociado a una alta incidencia de recurrencia y con un riesgo significativo de malignidades, sobre todo del tracto gastrointestinal, mama, órganos genitales y páncreas, por lo que debe efectuarse un estrecho seguimiento del paciente durante toda su vida, examinando el tracto gastrointestinal y los órganos sólidos que son susceptibles de sufrir tumores malignos (6, 9, 10). La laparoscopia permite un adecuado acceso para explorar y tratar pequeños pólipos intestinales, evitando así la clásica laparotomía (6)
El pólipo hamartomatoso solitario tipo Peutz-Jeghers, con historia familiar negativa y sin hiperpigmentación muco-cutánea, de rara presentación, es considerado por algunos autores como una entidad separada del SPJ. En estos casos no hay riesgo de cáncer gastrointestinal y por lo tanto no es necesario un seguimiento de alto riesgo (5, 11, 12).
|
|
|
|
Mujer de l9 años de edad, que fue internada por presentar náuseas, vómitos de tipo bilioentéricos, distensión abdominal a predominio en hemiabdomen inferior, falta de eliminación de gases y materia fecal, de l2hs de evolución, y dolor abdominal tipo cólico en cuadrante inferior derecho de abdomen. Al exámen físico se observaron manchas hiperpigmentarias lentiginosas en el rostro y periorificiales (boca y nariz) (Figuras 1 y 2). Resto del cuerpo sin particularidades.
Se instauró tratamiento médico con hidratación parenteral, sonda nasogástrica y observación durante 6 horas, con mala evolución. La Radiografía directa de abdomen, de pié y en decúbito dorsal, demostró signos de oclusión intestinal baja. Se realizó seriado contrastado con material iodado, de tubo digestivo alto, objetivándose a los 6 metros aproximadamente un stop del mismo en hemiabdomen inferior derecho (Figura 3).
La tomografía abdominal con doble contraste (oral y endovenoso) mostró una imagen sospechosa de intususcepción intestinal (imagen de doble contrasteen escarapela) en fosa iliaca derecha.
Se efectuó laparotomía exploradora observándose una invaginación intestinal ileo-ileal con compromiso vascular del segmento invaginado (Figura 4). Ante la apertura de este, se visualizó una tumoración exofítica en la luz del íleon, de unos 4 cm de diámetro, que actuó como motivo de la intususcepción. Se llevó a cabo una resección intestinal segmentaria y anastomosis enteroentérica término-terminal, con buena evolución y retransitación intestinal, dándose de alta a la paciente a los cinco días del postoperatorio.Anatomía Patológica: Se estudió un segmento de intestino delgado 70 cm de longitud (después de fijado), que se recibió abierto y sin contenido alimentario (lavado). La serosa estaba muy congestiva y sin placas de exudado purulento.
A 12 cm del límite quirúrgico más cercano, se observó una formación poliposa de 3.8 cm de diámetro, rojo-vinosa, de franco aspecto hemorrágico al corte, con un un pedículo mucoso de implantación de 2.5 cm de largo, sin evidencias macroscópicas de procesos tumorales infiltrativos (Figura 5). A 15 cm de esta formación poliposa y abarcando una extensión de 20 cm, la pared intestinal se veía muy edematosa y rojiza. El resto de la pieza operatoria, incluyendo límites quirúrgicos, no presentaba más formaciones tumorales ni otras lesiones de consideración.
Las secciones histológicas mostraron un pólipo constituido por glándulas de tipo intestinal, dispuestas en grupos irregulares, con corion inflamado no fibrótico, circundados por haces de fibras musculares lisas de variable espesor (Figuras 6 a 8). En sectores se observaba necrosis de coagulación y hemorragia, con desorganización de la disposición lobular en las glándulas vecinas y cambios citoestructurales que se interpretaron como regenerativos-reactivos, no habiendo evidencias de infiltración neoplásica del pedículo poliposo ni del resto de la pared intestinal. El área intestinal de 20 cm descripta en la macroscopia, tenía también necrosis y hemorragia limitada a la mucosa (infarto mucoso). El diagnóstico final fue de Pólipo Hamartomatoso Tipo Peutz-Jeghers, complicado con invaginacion intestinal (intususcepción) e infarto hemorragico del propio pólipo y de una porcion del segmento intestinal resecado.
|
|
|
|
El pólipo tipo Peutz-Jeghers debuta frecuentemente como una urgencia quirúrgica, ocasionando un verdadero abdomen agudo, que en nuestro caso se produjo por una obstrucción intestinal debida a una invaginación intestinal, la que a su vez llevó a un infarto de intestino y del propio pólipo. El riesgo de ser portador de estos pólipos no termina en el proceso abdominal agudo, ya que cuando el la poliposis intestinal y la hiperpigmentación mucocutánea se asocian a los antecedentes familiares, constituyendo el SPJ, aumentan a largo plazo las probabilidades de desarrollar malignidades intestinales y de órganos sólidos. La paciente no regresó para control después del alta médica definitiva, por lo que no pudimos constatar si algún familiar directo de ella era portador de pólipos intestinales, manchas lentiginosa muco-cutáneas o si había sido intervenido en algún momento por un cuadro de abdomen agudo. A pesar de esto, creemos que nuestro caso cae en la categoría de SPJ, ya que el pólipo intestinal que hallamos se acompañaba, al contrario de lo que sucede con el pólipo tipo Peutz-Jeghers solitario, de acentuada lentiginosis cutánea facial y periorificial, lo que nos sugiere que esos antecedentes familiares que no se pudieron determinar en su momento tienen que existir. Esto hace que la paciente deba ser controlada por toda su vida, ante el riesgo de recurrencias y malignidades.La anatomía patológica del pólipo tipo Peutz-Jeghers se basa en la presencia de glándulas hamartomatosas separadas en grupos irregulares por haces ramificados de la muscular de la mucosa. Encontramos áreas glandulares desorganizadas y con cambios reactivos, debido a que eran vecinas a las zonas infartadas del pólipo. De todas formas, nada nos indicó la presencia de verdaderos cambios displásicos, precursores de un desarrollo carcinomatoso en el seno del pólipo.
|
|
|
|
|
|
|
|
1. Leaper D : Tumours of the Small Intestine. In: Schwartz S., Ellis H, Maingot¨s; editors. Abdominal Operations. 9a ed.
|
|
|
|
- Javier Muñoz Moreno (03/10/2005 13:44:23)
- ELSIE BEATRIZ PICOTT RANGEL (13/10/2005 20:26:38)
- Mario Miguel Morales Wong (21/10/2005 0:49:32)
|
|
|
|
|
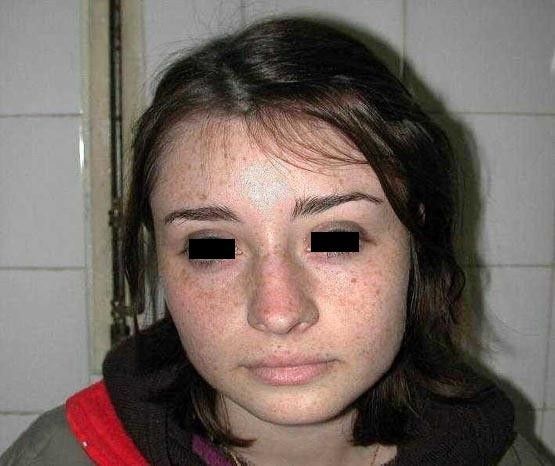 fiogf49gjkf0dFig. 1. Numerosas manchas hiperpigmentarias lentiginosas en el rostro y periorificiales (boca y nariz)">
fiogf49gjkf0dFig. 1. Numerosas manchas hiperpigmentarias lentiginosas en el rostro y periorificiales (boca y nariz)">
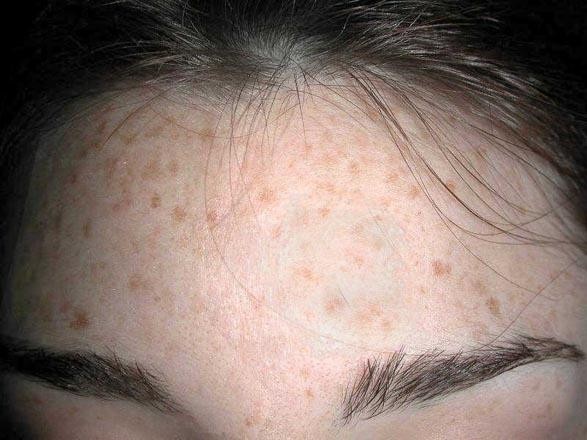 fiogf49gjkf0dFig. 2. Con más aproximación, zona frontal.">
fiogf49gjkf0dFig. 2. Con más aproximación, zona frontal.">
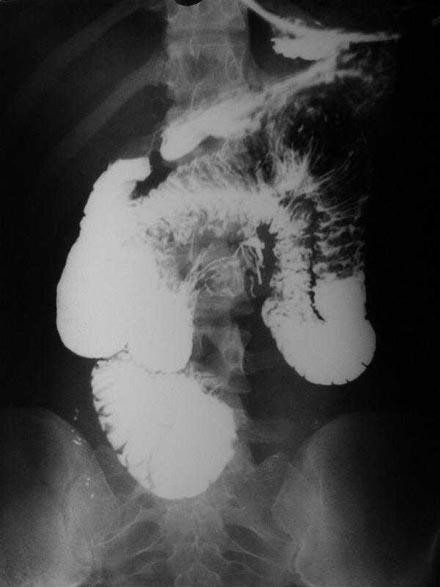 fiogf49gjkf0dFig. 3. Seriado contrastado con material iodado, de tubo digestivo alto, que muestra un stop del mismo en hemiabdomen inferior derecho.">
fiogf49gjkf0dFig. 3. Seriado contrastado con material iodado, de tubo digestivo alto, que muestra un stop del mismo en hemiabdomen inferior derecho.">
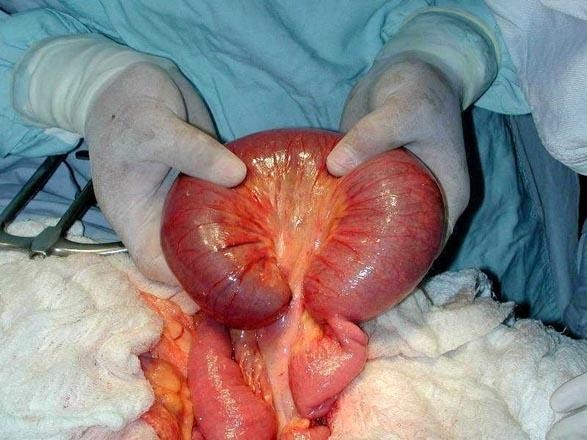 fiogf49gjkf0dFig. 4. Laparotomía exploradora que muestra el compromiso vascular del segmento invaginado.">
fiogf49gjkf0dFig. 4. Laparotomía exploradora que muestra el compromiso vascular del segmento invaginado.">
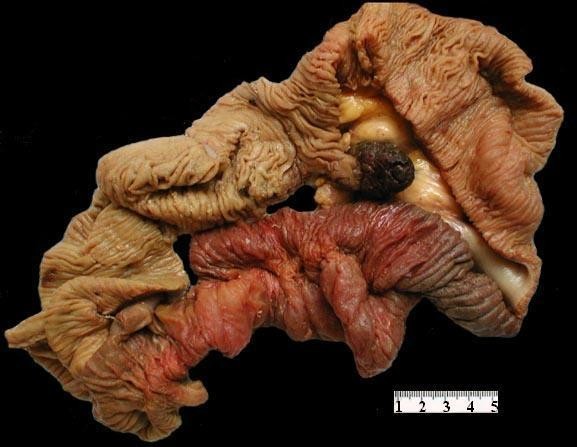 fiogf49gjkf0dFig. 5. Segmento intestinal resecado que incluye una formación poliposa de 3.8 cm de diámetro, rojo-vinosa, con un un pedículo mucoso de implantación de 2.5 cm de largo.">
fiogf49gjkf0dFig. 5. Segmento intestinal resecado que incluye una formación poliposa de 3.8 cm de diámetro, rojo-vinosa, con un un pedículo mucoso de implantación de 2.5 cm de largo.">
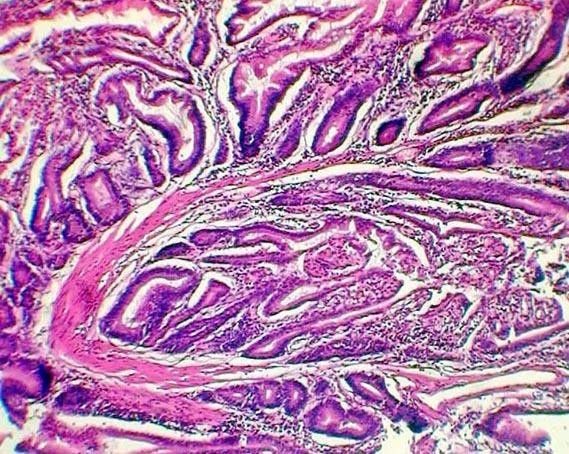 fiogf49gjkf0dFig. 6">
fiogf49gjkf0dFig. 6">
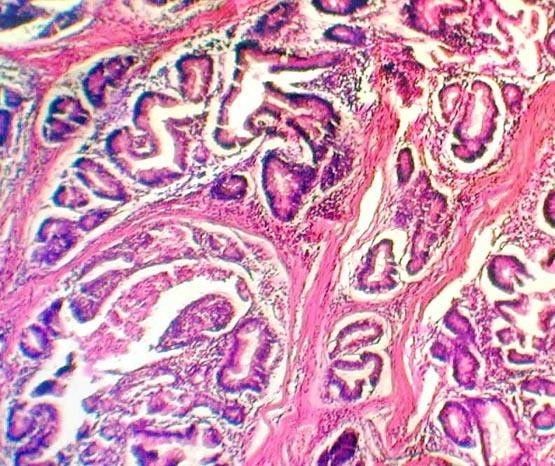 fiogf49gjkf0dFig. 7">
fiogf49gjkf0dFig. 7">
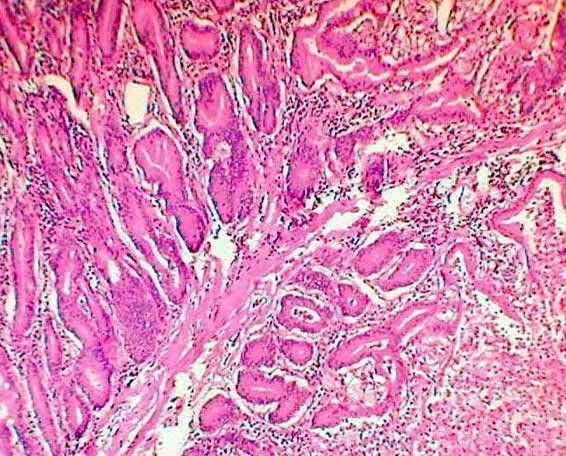 fiogf49gjkf0dFig. 8
Figs. 6 a 8. Glándulas hamartomatosas agrupadas en forma irregular por haces de fibras musculares lisas.">
fiogf49gjkf0dFig. 8
Figs. 6 a 8. Glándulas hamartomatosas agrupadas en forma irregular por haces de fibras musculares lisas.">
Web mantenido y actualizado por el Servicio de informática uclm. Modificado: 16/06/2015 15:10:50