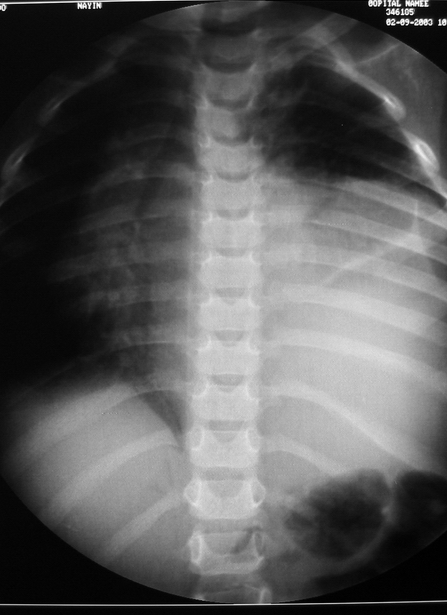
El blastoma pleuropulmonar (BPP) es un tumor pulmonar raro (
7,
9,
10), característico de la infancia, generalmente antes de los 6 años (
1,
3). También se han descrito casos en adultos, aunque se consideran entidades diferentes (
2,
3,
12). Clínicamente los BPP debutan con sintomatología inespecífica como: dificultad respiratoria, dolor torácico, anorexia o fiebre, simulando quistes infectados o una malformación quística congénita, especialmente la malformación adenomatoide quística congénita tipo 4 (
3,
4,
6,
7,
9). Metastatizan frecuentemente a cerebro, hueso, ganglios linfáticos, páncreas, riñones y glándulas suprarrenales (
3). Tienen un pronóstico desfavorable.
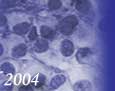
Histológicamente están constituidos por tejido mesenquimal maligno de aspecto embrionario o blastematoso, un estroma sarcomatoso y un componente epitelial atrapado, no son tumores bifásicos epiteliales y estromales como los de adultos (
6). Pueden afectar a pulmón, pleura y mediastino (
7,
9). En el componente sarcomatoso es frecuente la diferenciación rabdomioblástica, condroide y células similares a los fibroblastos (
1,
6).
El diagnóstico diferencial se realiza con otros tumores de células redondas y pequeñas. El linfoma y el neuroblastoma tienen diferente histopatología e inmunohistoquimica (LCA/ Proteína S-100, neurofilamentos, cromogranina, sinaptofisina) (
7,
8). La familia de tumores de Sarcoma de Ewing/Tumor Neuroectodermico Primitivo (TNEP) expresan fuerte inmunorreactividad de membrana con el marcador inmunohistoquímico O13 (HBA 71; p30/32 MIC 2; CD 99), que aunque no es patonogmónico (rabdomiosarcoma embrionario, otros sarcomas de partes blandas y linfoma linfoblástico) es característico. El rabdomiosarcoma está constituido por masas sólidas de células pequeñas y basofílicas, carece de áreas blastematosas y de diferenciación de otro tipo (condroide o fibroblástica) (
7,
9). Fibro-leiomiosarcoma con patrón sarcomatoso de células fusiformes, es más compacto y carece de pleomorfismo celular. El tumor de Askin presenta un diagnóstico diferencial más difícil, carece de la diferenciación a cartílago, tejido fibroso o rabdomioblastos (
9).
Los estudios inmunohistoquímicos ayudan al diagnóstico, sin embargo, la presencia de las áreas blastematosas y la diferenciación condroide (más frecuente) o rabdomioblastica son claves en el diagnóstico.
En la actualidad todos reciben el nombre de blastoma pleuropulmonar (BPP), aunque presentan un ámplio espectro morfológico, desde formas quísticas hasta sólidas. Se subdividen en tipo I (quístico), tipo II (mixtos) y tipo III (de predominio sólido) (
6,
9). Pueden comprometer a pulmón, pleura, pericardio y mediastino.
En cuanto al tratamiento, dada su rareza no hay estudios concluyentes, la cirugía, la poliquimioterapia y en algunos casos la radioterapia (cuando la extirpación no es total
6) juegan un papel muy importante, sin embargo, no previenen la recurrencia local, ni las metástasis. En general las formas sólidas presentan peor pronóstico que las quísticas (
7,
9). La supervivencia está en torno al 45-50% a los 5 años (
7). Es importante unificar criterios diagnósticos, determinar su prevalencia y comportamiento biológico en el tratamiento, ya que son enfermedades muy poco frecuentes (
13).
Debemos considerar en el futuro el estudio genético de los pacientes y sus familiares, a fin de detectar alteraciones cromosómicas que favorezcan el desarrollo de estas neoplasias o similares (
11) En este sentido se está investigando la asociación de BPP con la pérdida de heterocigosidad detectada en algunos familiares en 11p15.5 (
6).









 Zoom
Zoom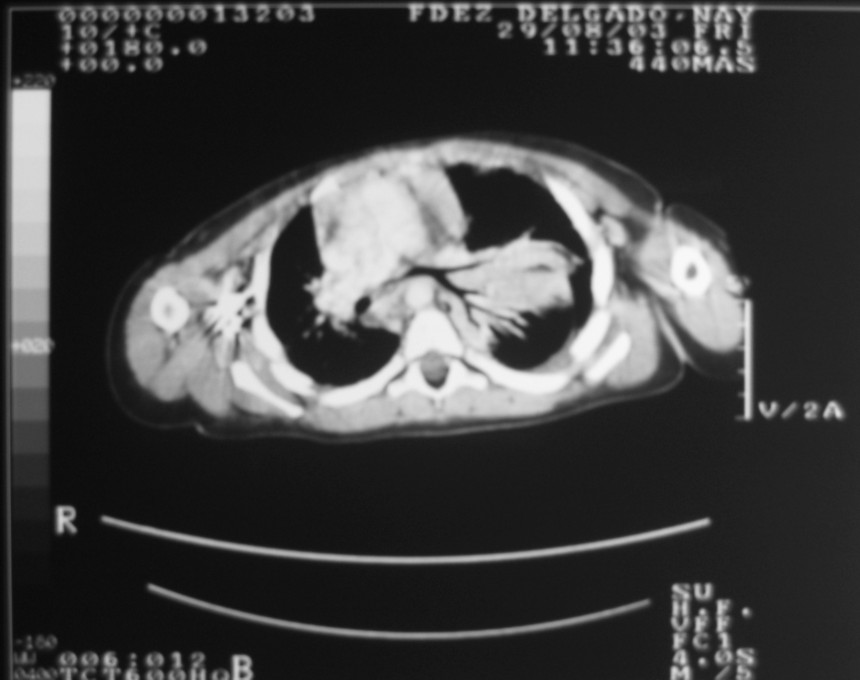 Zoom
Zoom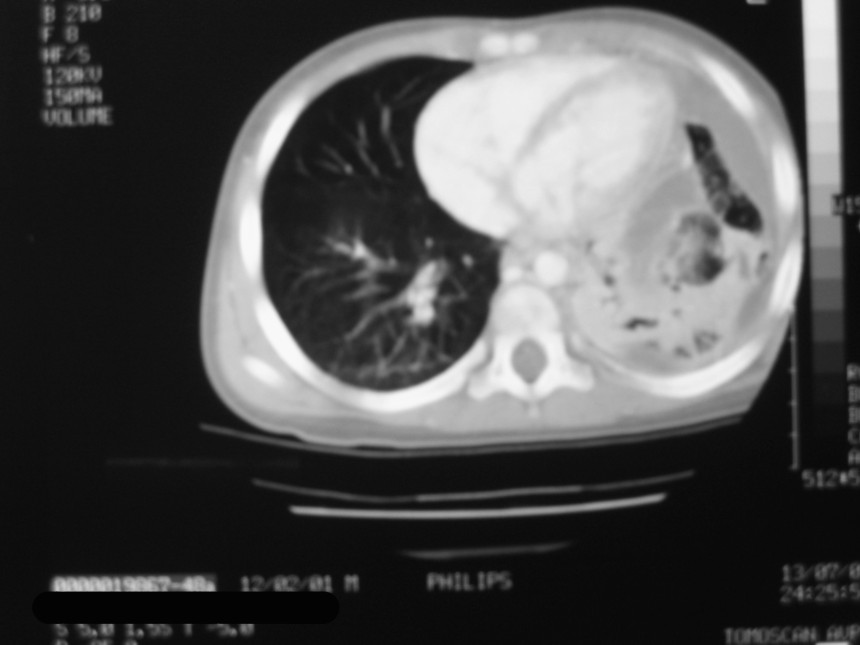 Zoom
Zoom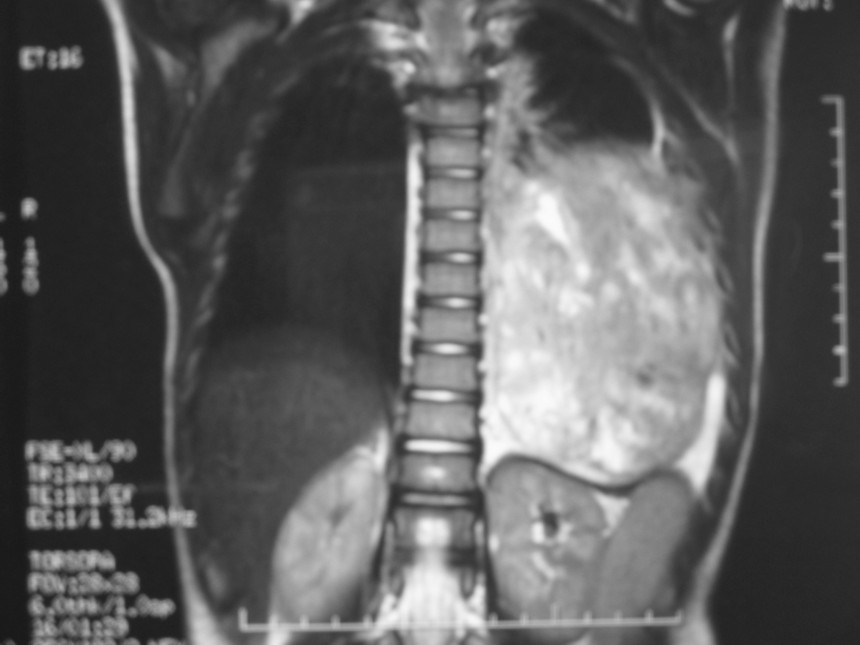 Zoom
Zoom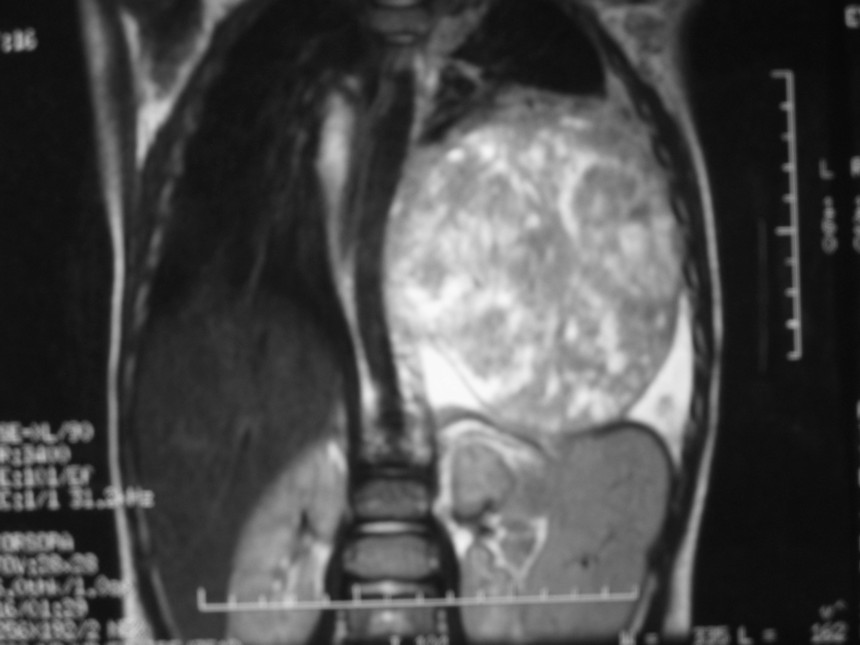 Zoom
Zoom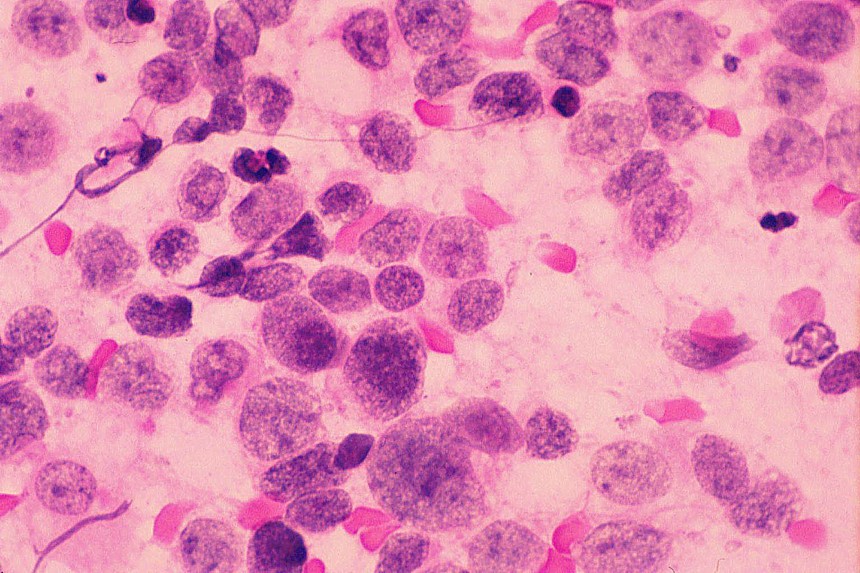 Zoom
Zoom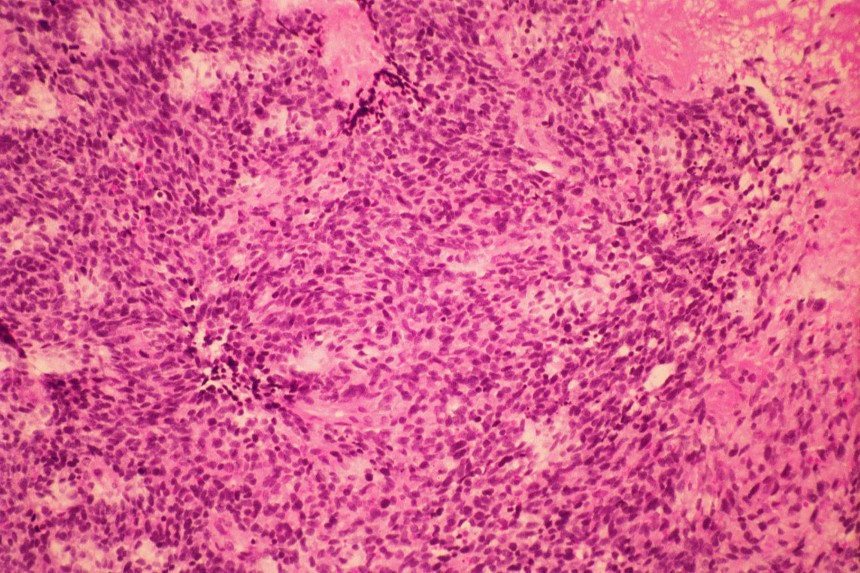 Zoom
Zoom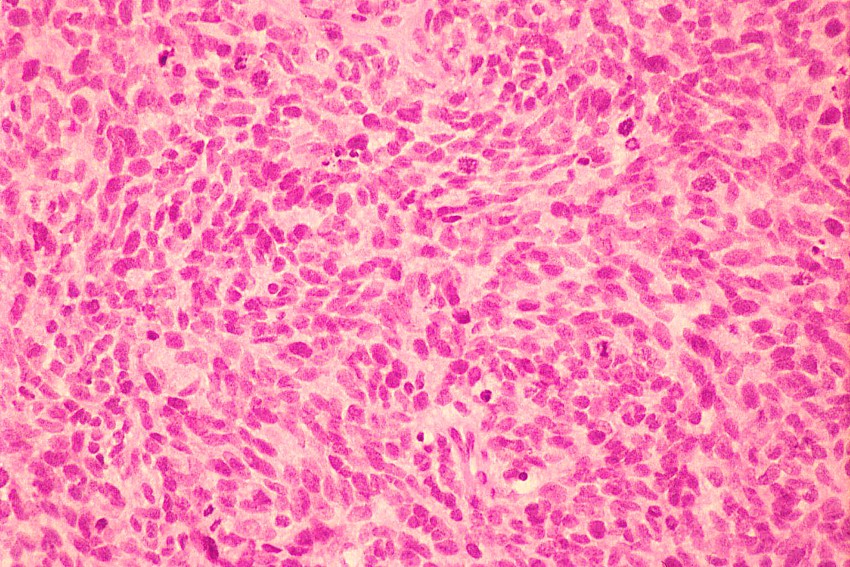 Zoom
Zoom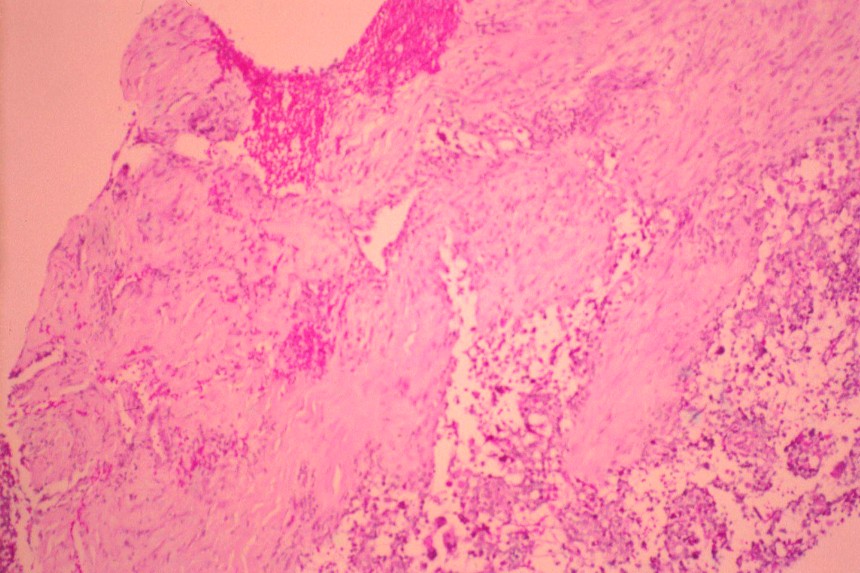 Zoom
Zoom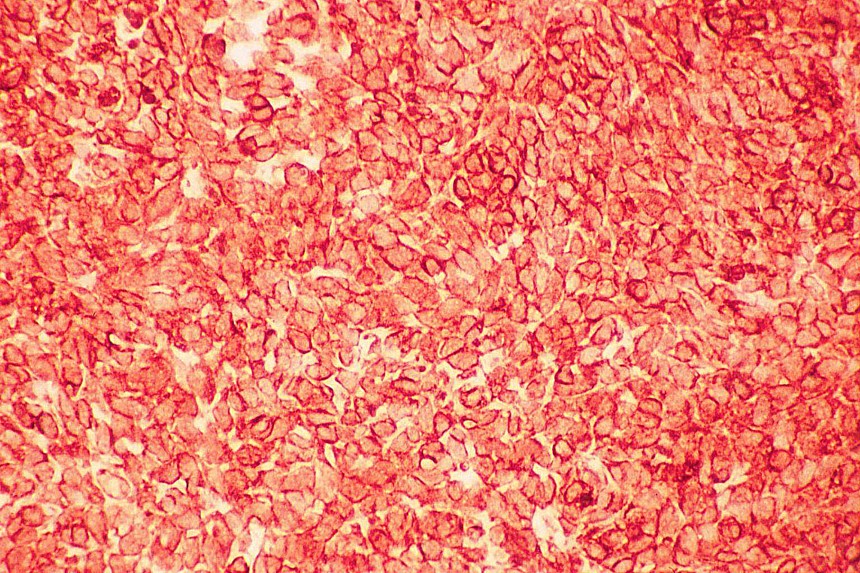 Zoom
Zoom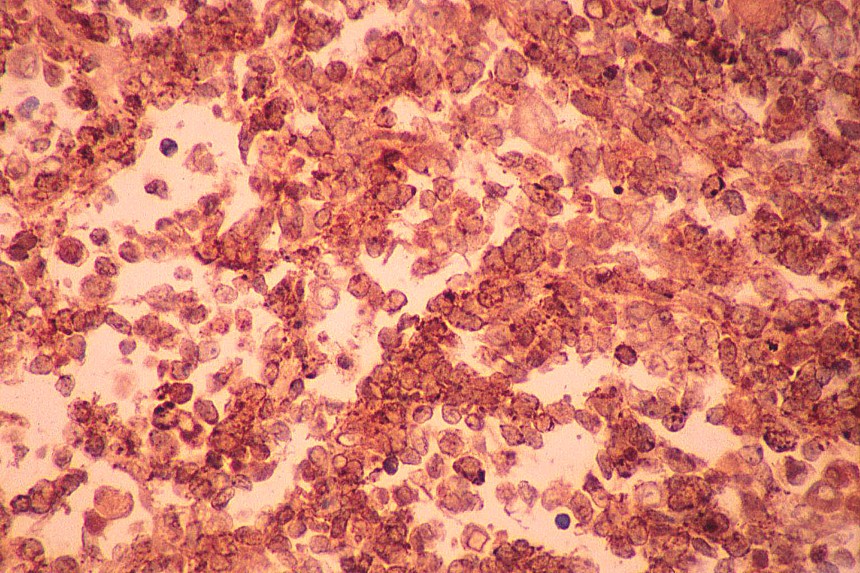 Zoom
Zoom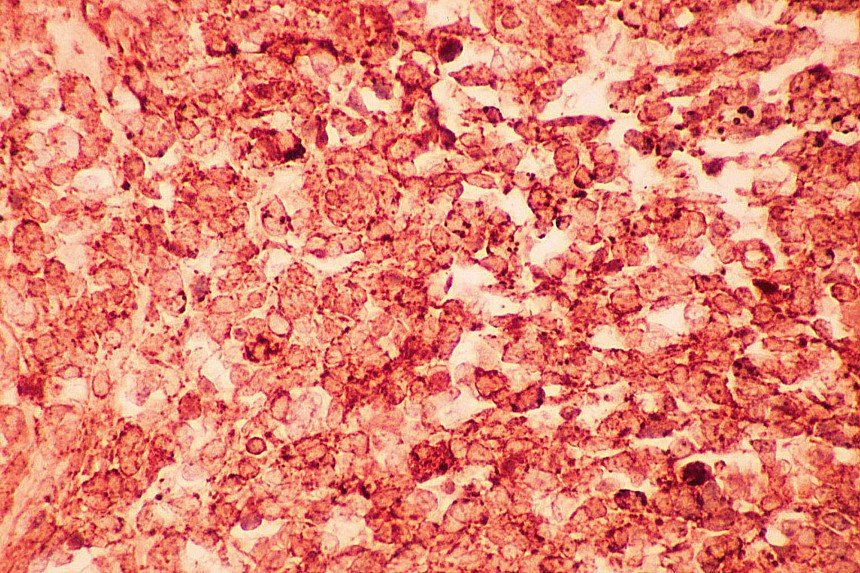 Zoom
Zoom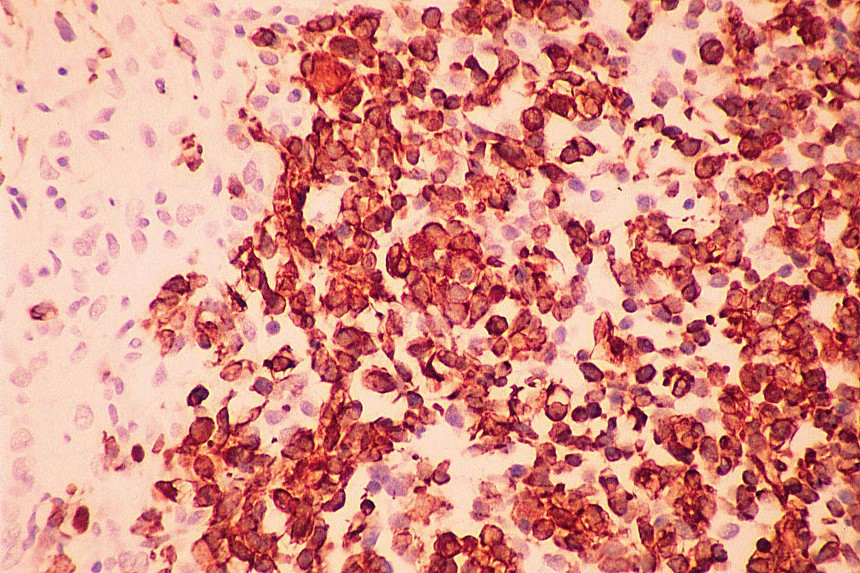 Zoom
Zoom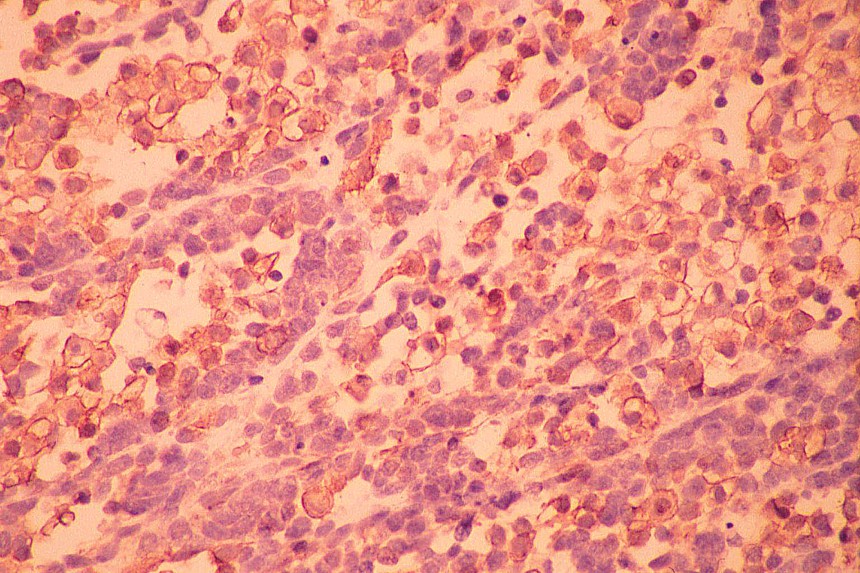 Zoom
Zoom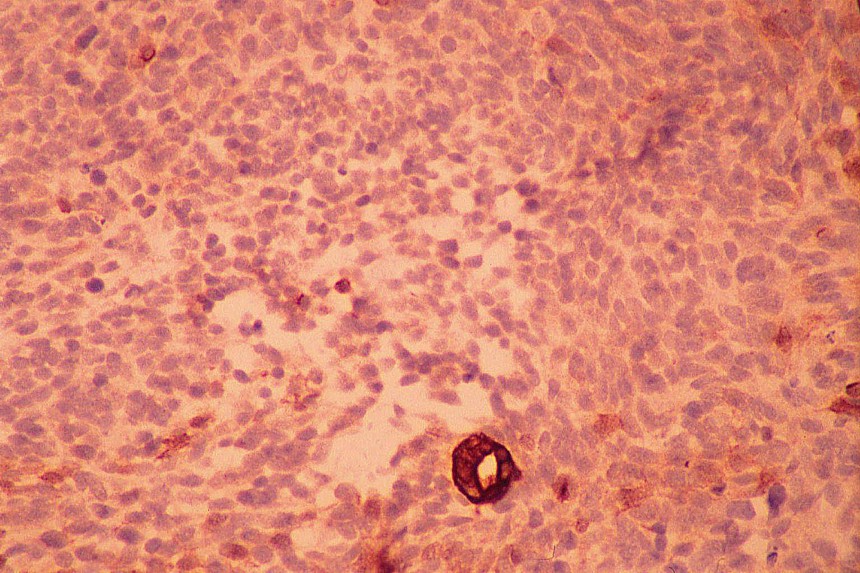 Zoom
Zoom