La nesidioblastosis es reconocida en la actualidad como la causa más importante de hipoglicemia-hiperinsulinémica persistente en el recién nacido y el lactante(
3),(
4), por lo que a pesar de ser un trastorno endocrino poco frecuente, debe sospecharse ante un lactante o recién nacido con historia temprana de hipoglicemias severas persistentes y resistentes al tratamiento Desde el punto de vista clínico son características además las crisis hipoglicémicas agudas e intensas, prolongadas y rebeldes al tratamiento, así como gran intranquilidad, palidez, sudación, apatía, irritabilidad, rechazo al alimento y convulsiones, pudiendo llegar al coma(
4). Algunos autores consideran que pueden existir formas subagudas que ocasionan muerte súbita en el recién nacido y el lactante.
El diagnóstico se realiza por el cuadro clínico, pruebas bioquímicas que evidencien hipoglicemia e hiperinsulinemia y por estudio anatomopatológico, especialmente con técnicas de inmunohistoquímica de cuyos resultados existen varios reportes en la literatura (
2),(
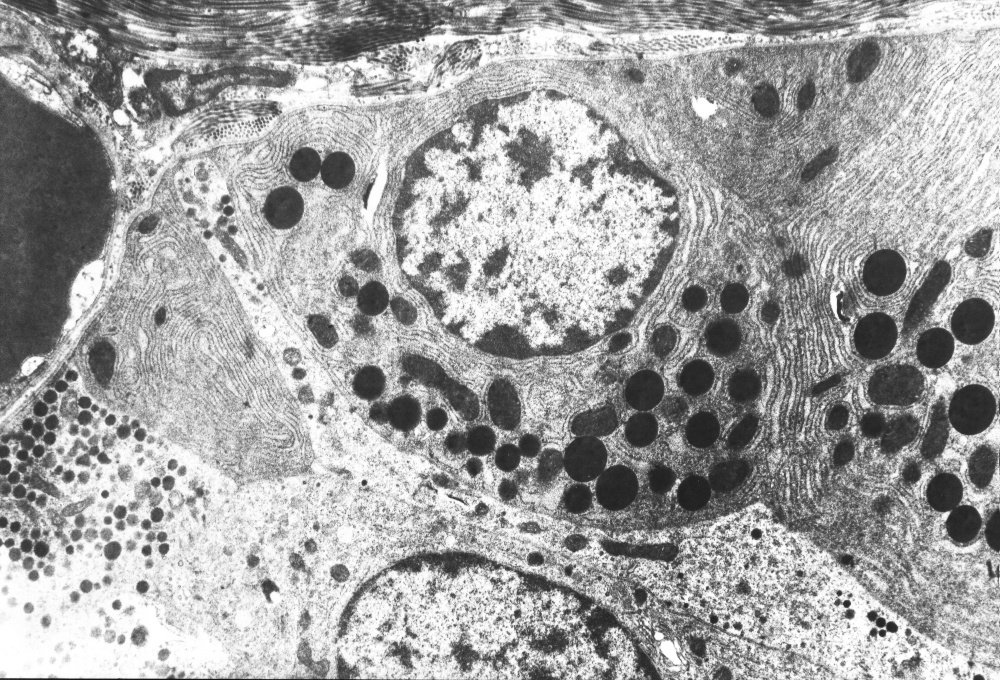
5) . Diversos autores coinciden en plantear que el hallazgo morfológico más importante consiste en la observación de células endocrinas fuera de su localización habitual a nivel de los islotes, las que se distribuyen difusamente entre las células ce los acinos y conductos pancreáticos, comprobándose con técnicas de inmunohistoquímica que estas células secretan insulina y con los estudios ultra estructurales su naturaleza endocrina.
A pesar de que se han practicado diferentes modalidades de tratamiento médico, es compartida por la mayoría, la opinión que el tratamiento de elección es la pancreatectomía subtotal (aproximadamente el 90 %), pero existen notificaciones de casos donde ha sido necesario una pancreatectomía total (
6),(
7). A los dos casos presentados por nosotros se les realizó pancreatectomía subtotal con buenos resultados.
A nuestros casos se les realizaron estudios inmunohistoquímicos y de microscopía electrónica, siendo nuestros hallazgos distintivos y diagnósticos de la entidad, por lo que coinciden con los obtenidos por otros autores(
5),(
8). En nuestra experiencia el estudio biópsico se realizó por el método convencional de inclusión en parafina, para la microscopía óptica y la inmunohistoquímica y no se realizaron cortes por congelación. Sin embargo existen comunicaciones científicas (9) donde se valora la realización de biopsias por congelación con vistas a adoptar un protagonismo activo en la decisión del tipo de pancratectomía a realizar, aunque, otros científicos lo consideran de difícil evaluación, opinión esta compartida con nosotros.
La patogenia de esta enfermedad aún no ha sido completamente dilucidada aunque existen demostraciones mediante estudios genéticos y moleculares, de la existencia de mutaciones de los genes que codifican el receptor de las sulfonilurea así como en los casos familiares, de pérdida materna del gen localizado en el cromosoma 11 (11p15), demostrándose además en numerosos pacientes la ausencia del ATP relacionado con la función de los canales de potasio (
3),(
10).
Publicaciones más recientes a partir de los trabajos de Rahier y Sempoux en 1998, sugieren referirse a la lesión morfológica como nesidiodisplasia de tipo focal o difusa (11,12,13) de acuerdo a la teoría de una falla en el desarrollo del páncreas endocrino
considerando además a la nesidioblastosis incluida en las de tipo difusa.
Por último, consideramos esta entidad como sumamente interesante en el universo de la patología endocrina y pediátrica cuyo tratamiento eficaz y oportuno depende del trabajo de un equipo multidisciplinario dónde se incluye el patólogo.
Conclusiones:
A pesar de que la nesidioblastosis es un trastorno endocrino poco frecuente, debe sospecharse ante un lactante o recién nacido con historia temprana de hipoglicemias severas persistentes y resistentes al tratamiento.
Los estudios anatomopatológicos especialmente los ultra estructurales e inmunohistoquímicos son de vital importancia para corroborar el diagnóstico.
El incremento en el área total ocupada por células endocrinas secretoras de insulina, es el hallazgo morfológico más importante obtenido por nosotros, lo cual coincide con lo reportado por otros autores.









 Zoom
Zoom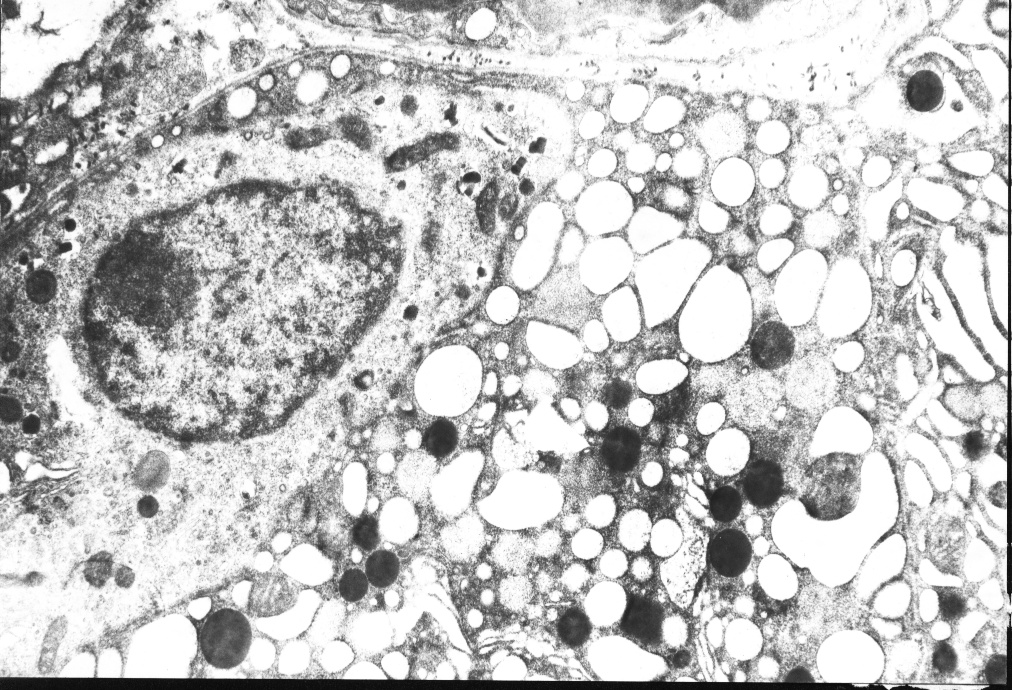 Zoom
Zoom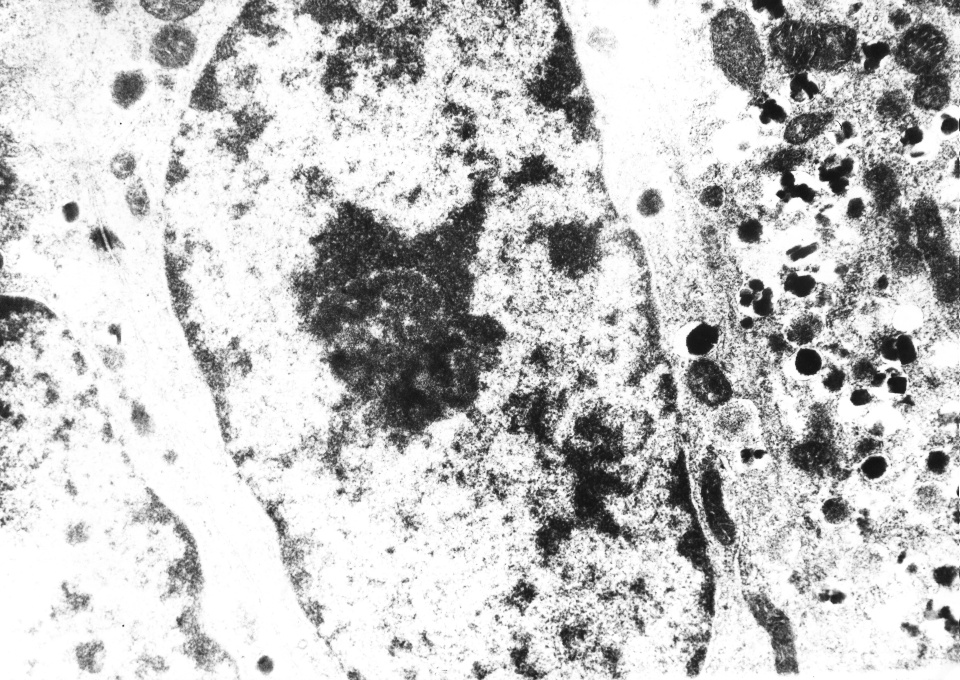 Zoom
Zoom