|
El Schwannoma Maligno (SM) es la principal malignidad de nervios periféricos, ocupa el 10% de todos los sarcomas de tejidos blandos y está considerado el más esquivo y pobremente definido los mismos. (1)
Las localizaciones más frecuentes en orden decreciente son: Miembros inferiores, miembros superiores, retroperitoneo, tórax, cabeza y cuello. Independientemente de las localizaciones que se citan, aproximadamente la mitad de los SM no se desarrollan a partir de troncos nerviosos visibles macroscópicamente, lo cual dificulta su diagnóstico. (1, 2)
El SM ocurre en menos de la mitad o la tercera parte aproximadamente, en pacientes portadores de una neurofribromatosis tipo 1 (NF1) o Enfermedad de Von Recklinghausen, luego de un período de latencia prolongado de 10 a 20 años. Las variables de la edad y el sexo varían según esta incidencia, pues se cita que el SM ocurre en adultos entre los 20 y 50 años de edad, excepto en los pacientes con NF1, donde se presenta en más temprana edad. Asimismo se cita que la incidencia es igual para ambos sexos, pero en los casos con NF1 predomina el masculino. Lar raza escapa de estas relaciones y se observa en todas por igual.
Entre los factores etiológicos la asociación con la NF1, se considera la de mayor peso. No obstante, se cita además la radioterapia con intervalo entre la radiación y la terapia de más de 4 años y los carcinógenos químicos en el medio. Esta carcinogénesis está fundamentada por los resultados de la patología experimental. Estos tumores pueden ser inducidos en animales de laboratorio por inyección trasplacentaria de etilnitrosourea y por la administración de metilcolantreno (1)
El SM es un tumor que se caracteriza por sus asociaciones y aunque la que se cita preferentemente es la NF1, se reporta la asociación a numerosas neoplasias tales como: Rabdomiosarcomas, liposarcomas, gliomas del nervio óptico, astrocitomas de hemisferios cerebrales o cerebelosos, astrocitomas medulares, meningiomas, feocromocitomas, nefroblastomas, leucemias, etc... Quizás la asociación con los tumores anteriormente relacionados esté basado en la primera condición citada, pues la NF 1 es una variedad de facomatosis que se caracteriza por síndromes neurocutáneos con hamartomas y tumores diseminados por todo el cuerpo, pero más con afectación del sistema nervioso y piel. El gen implicado en la NF 1 está situado en el 17 q 11.2. Es una de las enfermedades genéticas más frecuentes, afecta una de c/3000 personas. Se trata de un trastorno autosómico dominante de alto grado de penetración caracterizado por neurofibromas, gliomas del nervio óptico, nódulos pigmentados del iris (Nódulos de Lisch) y manchas hiperpigmentadas cutáneas (Manchas de café con leche). (1-2)
Se plantea que el gen de la NF 1 actúa realmente como un gen supresor tumoral y codifica una proteína, la neurofibromina, que se expresa ampliamente y con mayores niveles en el tejido neural. Se describen Síndromes familiares como la NF 1 y la neurofibromatosis tipo 2, la esclerosis tuberosa, la Enfermedad de Von Hipel Lindau y Síndromes esporádicos como el Sturge Weber.
En nuestro caso únicamente corroboramos la asociación de un paciente portador de una NF1 con un SM. El paciente refería: Ser el primer miembro afectado en la familia, que solamente padecía de los neurofibromas diseminados por todo el cuerpo desde la niñez y que nunca le habían causado problemas, pues él se consideraba muy saludable.
Por el cuadro clínico, consideramos que la aparición del SM no fue una progresión de un neurofibroma con transformación maligna, tal como se cita entre un 3 a un 13 % sobretodo en los plexiformes, pues según refiere el mismo paciente, le apareció esa masa en la axila izquierda 4 meses antes, la cual tuvo un crecimiento sumamente rápido al igual que se cita en la literatura.
Al examen microscópico llamó la atención que en muy pocas áreas las células tumorales recordaban las típicas células de Schawnn, con núcleos alargados y extensiones bipolares prominentes. Además no se observó la característica formación de empalizada, aunque sabemos que esto en ningún momento debe interpretarse como un dato patognomónico de SM, ni siquiera de tumor neurogénico. Sin embargo, entre las células con límites mal definidos se observaban bandas de colágeno, que sí solas, son aspecto poco comunes del SM, junto a los nódulos hialinizados son distintivas del mismo.
Dada la presencia de las células epitelioides en la neoplasia y más aún con la ausencia de datos clínicos, se valoró como otra posibilidad el diagnóstico de un Sarcoma Sinovial o como diagnóstico diferencial sumamente importante que se cita, además del fibrosarcoma y el leiomiosarcoma. (1-3).
La presencia de estructuras glandulares glandulares en un sarcoma monofásico debe hacer pensar en un sarcoma sinovial bifásico o en un sarcoma neural con componente glandular (3)
La clínica del enfermo, al carecer de antecedentes malformativos neurales orientaron al diagnóstico de un sarcoma sinovial (4). No obstante, después de conocer que el paciente era portador de una NF 1 y facilitarse nuestro diagnóstico de SM basado fundamentalmente en la morfología y la asociación de esta neoplasia con dicha entidad, buscamos la asociación del Sarcoma Sinovial con la neurofibromatosis y no encontramos reportes bibliográficos (1-6).
Las 3 variedades histológicas del SM que se citan son: El SM con diferenciación rabdomioblástica (Tumor Maligno de Tritón), el SM glandular y el SM epitelioide (1,2, 5).
El SM epiteliode es una variedad histológica poco usual, aproximadamente el 5% de todos estos tumores. Se observa principalmente en nervios mayores y su histología es muy variable. El aspecto más característico es cordones de grandes células epiteliodes, lo cual coincide con nuestro caso, no así las citas en ocasiones de diversos cuadros morfológicos semejantes al melanoma y también patrón nodular e incluso formación de rosetas. El tejido mixoide es escaso y entremezclado, tal como se observó en nuestro caso. No detectamos focos aislados de osteoide, cartílago, ni calcificaciones.
Se considera que el origen en un nervio es imprescindible para poder establecer el diagnóstico de SM, y si bien en nuestro caso no pudimos realizarlo en el estudio macroscópico, en los cortes histológicos se detectó el origen en los mismos.
La marcada infiltración neoplásica que se observó hacia la periferia en el tejido fibroadiposo, así como los numerosos ganglios linfáticos con depleción linfocítica y marcada infiltración grasa, tradujeron la baja inmunidad celular de este paciente, como respuesta del huésped a la neoplasia, lo cual se correlaciona con la fatal evolución del mismo.
La Inmunohistoquímica se cita variable y no muy precisa, relacionada preferentemente con la Proteína S-100 y la Queratina. (1,6)
En la microscopía electrónica los SM muestran axones en el citoplasma y una membrana basal alrededor de la membrana plasmática. Además de la lámina basal, la matriz extracelular contiene colágeno típico y espaciado.
En la variedad epitelioide, la escasez de casos no permite una afirmación significativa en cuanto al valor relativo de la microscopía electrónica en esta forma poco usual de este tumor. En 2 casos citados en la literatura las cálulas epiteliodes mostraban pocos aspectos distintivos y no se parecían a las células de Shawnn (1). Sin embargo, en un 3er. Caso (7), las células epiteliodes se identificaron fácilmente como células de Shawnn, debido a numerosas extensiones citoplasmáticas cubiertas por lámina basal. Estos hallazgos se consideran compatibles, pues hay ciertas evidencias de que las células fibroblásticas pueden participar en los SM con NF 1. Se cita que el fibroblasto perineural y las células de Schawnn pueden tener modulaciones estructurales por su ubicación en las vainas nerviosas.
Según Lombart Bosch (8), independiente de la inmunohistoquímica y la microscopía electrónica, la Biología molecular es la única concluyente y definitiva, pero él mismo precisa que este procedimiento experimental no debe ser considerado como un sistema de diagnóstico clínico- asistencial (Tumores xenotransplantados en ratones atímicos).
A pesar del número limitado de casos informados de SM variedad histológica epitelioide, no hay duda de que son neoplasias altamente malignas y que deben ser tratadas de esta forma. Se cita que por lo menos la mitad de los pacientes informados, desarrollaron metástasis a distancia habitualmente a los pulmones. Sin embargo, aún no se sabe con certeza, si esta variante conlleva a un mayor riesgo de metástasis a ganglios linfáticos que el SM común.(1,2,5)
En nuestro caso coincide la evolución desfavorable de esta variante histológica. No obstante, a pesar de que el paciente falleció en un cuadro de Bronconeumonía Bacteriana que pudo tener de base una diseminación metastásica a pulmones, nos es imposible la corroboración de este hecho, ya que no se practicó la autopsia.
En general el pronóstico del SM se considera grave y más aún si el paciente es portador de una NF 1, pues el 80 % fallece a consecuencia del tumor, con una sobrevida de menos de 5 años. Las recidivas son frecuentes y la diseminación hemática principalmente es a pulmones y huesos. Hasta el momento ninguno de loa agentes quimioterápicos utilizados en el tratamiento paliativo han dado resultados alentadores (1,2,5)
En nuestro caso, la ausencia de datos clínicos en la solicitud de la biopsia pudo haber causado un gran error diagnóstico al carecer de una visión integral del enfermo-enfermedad, ya que los mismos se recuperaron de modo casual.
La adecuada correlación clínico- patológica y macro- microscópica contribuyeron a una mayor confiabilidad en el diagnóstico.
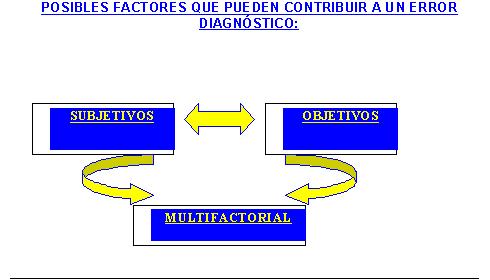
Se citan muchos trabajos en la literatura donde se insiste en las variables que pueden contribuir a un error diagnóstico, con la ilustración de numerosos casos (9) . En nuestro trabajo nosotros sintetizamos las mismas en los cuadros 1 y 2, y consideramos fundamental aplicar este algoritmo de acción en la metodología del procedimiento de la biopsia, a los médicos en formación y en especial a los patólogos.
Nuestras limitantes en los métodos especiales de la Anatomía Patológica, nos frenan en el campo de la investigación científica de nuestros diagnósticos biópsicos. No obstante, se demuestra la gran importancia de las variantes reflejadas anteriormente, en el procedimiento de la metodología correcta del diagnóstico biópsico, y sobretodo como esencia en la rutina de la labor asistencial. Los métodos investigativos en este caso, a pesar de lo elevado de los costos, no hubiesen resultado confiables, aunque en ningún momento menospreciamos su importancia en el desarrollo del campo científico- técnico-investigativo.
|









 Zoom
Zoom Zoom
Zoom Zoom
Zoom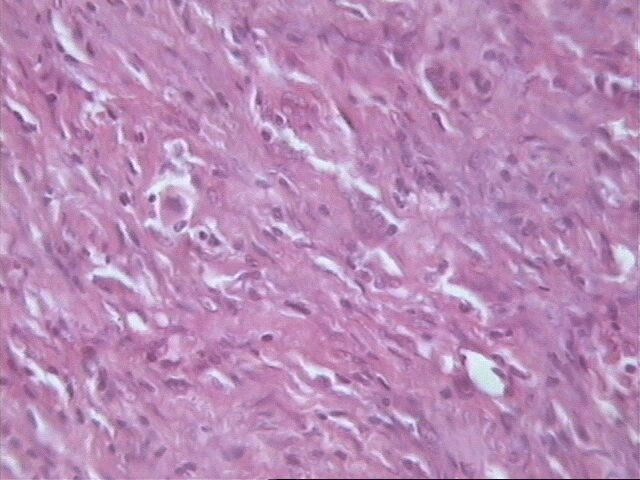 Zoom
Zoom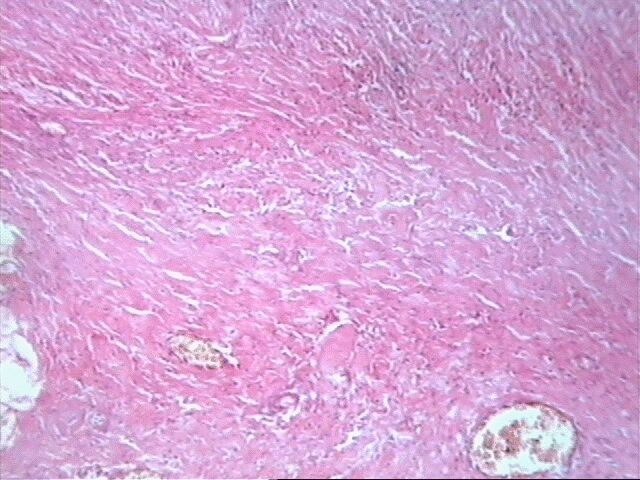 Zoom
Zoom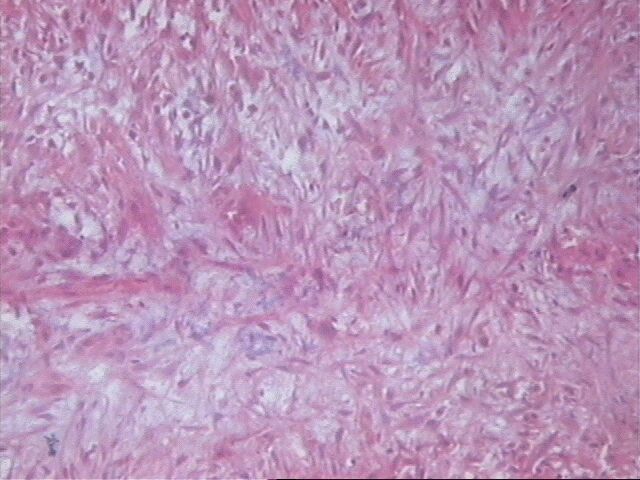 Zoom
Zoom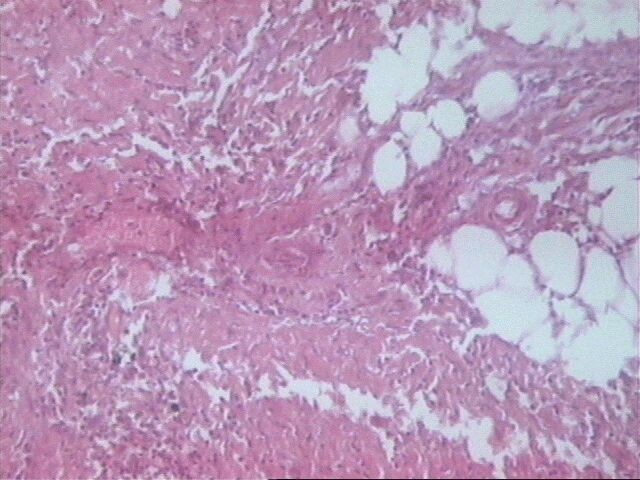 Zoom
Zoom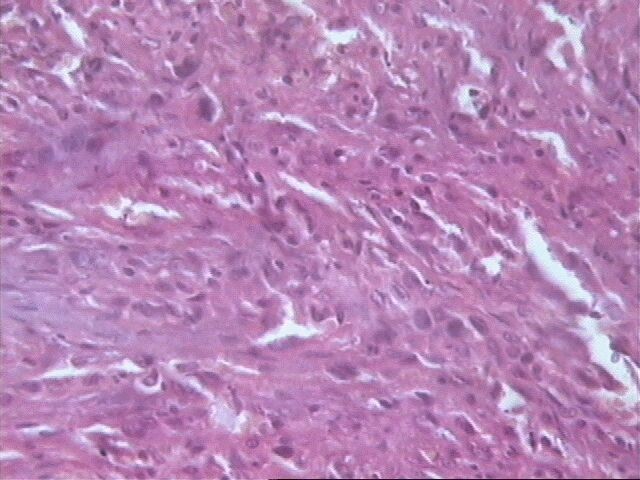 Zoom
Zoom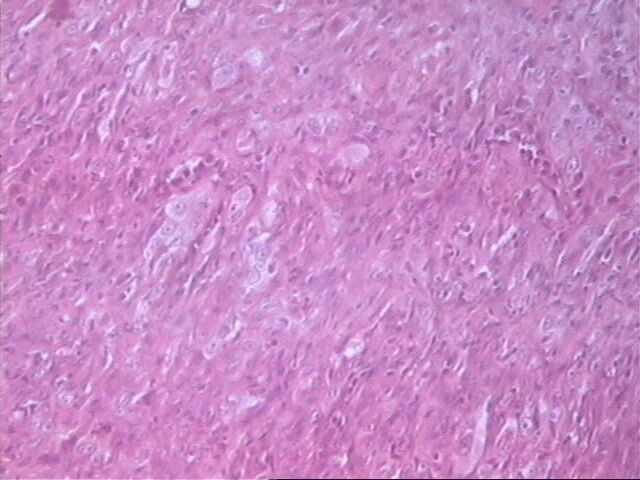 Zoom
Zoom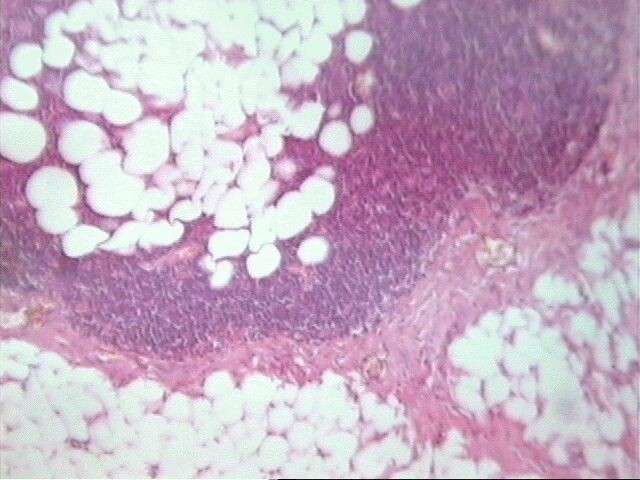 Zoom
Zoom