|
Resumen
buy abortion pill online can i buy abortion pill online INTRODUCCION:
La microlitiasis alveolar pulmonar es una rara entidad pulmonar, autonómica recesiva, caracterizada por la presencia de microlitiasis. Estos son pequeños corpúsculos constituidos por fosfato de calcio que se depositan en la luz alveolar.
Se ha reconocido la presencia de mutaciones a nivel del gen SLC34A2, responsable de dicha entidad. La incidencia es mayor en mujeres.
CASO CLÍNICO:
Hombre de 71 años que se presenta con un cuadro de insuficiencia respiratoria severa.
Con diagnóstico clínico radiológico de una enfermedad intersitcial difusa del pulmón se realiza cepillado y biopsia transbrónquica.
El estudio histológico mostró la presencia de cuerpos cálcicos laminados intraalveolares, asociados a fibrosis y escaso infiltrado inflamatorio mononuclear.
DISCUSIÓN:
Presentamos este caso por lo poco frecuente de esta entidad y por la forma en la cual se logró el diagnóstico haciendo especial mención al cepillado y biopsia trasbronquica como forma de diagnóstico.
En estos casos es importante establecer el diagnóstico diferencial con otras posibles causas de formación de corpúsculos intraalveolares.
|
|
Introducción
La microlitiasis alveolar pulmonar (PAM) es una rara enfermedad hereditaria, del pulmón, autosómica recesiva, caracterizada por la formación y depósito de cálculos de fosfato de calcio en la luz alveolar. Se han reportado aproximadamente 300 casos en la literatura. La exacta incidencia de esta entidad no se conoce. Se han reportado casos familiares. Recientemente se ha encontrado una mutación en el gen SLC34A2, responsable de esta enfermedad. La edad de diagnóstico de la entidad suele ser alrededor de los 40 años, aunque se han reportado casos desde la niñez hasta los 80 años. Existe un aumento de la incidencia en pacientes con ascendencia turca. La forma de presentación más frecuente es la asintomática. Otros se presentan con distinto grado de disnea, algunos con deterioro progresivo y acelerado de la función respiratoria y posterior muerte. Los pacientes presentan típicamente un defecto restrictivo de la función pulmonar. Se han reportado casos con asociación de litiasis renal y calcificaciones pleurales.
|
|
CASO CLÍNICO:
Hombre de 71 años que se presenta con un cuadro de insuficiencia respiratoria severa.
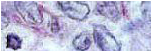
Con diagnóstico clínico radiológico de una enfermedad intersitcial difusa del pulmón se realiza cepillado y biopsia transbrónquica.
El estudio histológico mostró la presencia de cuerpos cálcicos laminados intraalveolares, asociados a fibrosis y escaso infiltrado inflamatorio mononuclear.

Cepillado broncoalveolar -

-

Cepillado broncoalveolar -

-

Biopsia transbrónquica -

-

-

-

-

-

-
|
|
DISCUSIÓN:
Presentamos este caso por ser una patología extremadamente rara, la cual se presentó en un paciente de 71 años sin elementos clínicos en la esfera respiratoria con anterioridad al cuadro actual. Como ya hemos visto la presentación más frecuente es la asintomática, siendo posible un rápido deterioro de la función respiratoria una vez iniciados los síntomas, muchos de los cuales culminan con la muerte del paciente. Esta parece ser la evolución de nuestro paciente, aunque el inicio de los síntomas fue más tardío.
Destacamos el valor diagnóstico del cepillado y biopsia transbrónquica en este paciente, ya que no fue planteado el diagnóstico con otras técnicas paraclínicas como la radiografía de tórax, la cual suele ser diagnóstica.
Por último debemos plantear y descartar los diagnósticos diferenciales, como la presencia de cuerpos amiláceos y calcificaciones alveolares por otras causas. La PAM puede diferenciarse de la presencia de cuerpos amiláceos por el mayor tamaño de los primeros y por la ausencia de calcificación en los segundos. Por otro lado los cuerpos amiláceos suelen tener un pequeño centro pigmentado que no se reconoce en la PAM.
Por otro lado la cantidad de microlitiasis alveolar la distingue de las distintas formas de calcificaciones del pulmón.
|
|
Bibliografía
1) Practical Pulmonary Pathology. Kevin O. Leslie, Mark R. Wick
2) Pulmonary alveolar microlithiasis: review and management. Tachibana T, Hagiwara K, Jonkoh T. Curr Opin Pulm Med. 2009 Sep; 15(5): 486-90
3) Pulmonary alveolar microlithiasis in a textil worker. Akyilidiz EU, Ursavas A. Ogur U. Ann Thorac Surg. 2009 Sep;88(3):e22-4.
4) Alveolar microlithiasis with severe interstitial fibrosis leading to lung transplantation. Coulibaly B, Fernandez C, Reynaud-Gaubert M, D`Journo X, Doddoli C, Taséi AM. Ann Pathol. 2009 Jun;29(3):241-4. Epub 2009 Jun 12.
5) Pulmonary alveolar microlithiasis: report of four cases. Marc K, Bourkadi JE, Jahid A, Cherradi N, Benamor J, Mahassini N, Fassy MT, Iraqi G. Rev Pneumol Clin. 2008 Oct;64(5):221-4. Epub 2008 Oct 26.
6) Pulmonary alveolar microlithiasis: clinical features, evolution of the phenotype, and review of the literature. Castellana G, Gentile M, Castellana R, Fiorente P, Lamorgese V. Am J Med Genet. 2002 Aug 1;111(2):220-4.
|

 COMUNICACIONES
COMUNICACIONES