|
Osteogénesis Imperfecta
Zulma S. Quintero N.[1], Isabel Guerra[2], Jose Javier Aguirre[2], Ainara Azueta[2]
(1) Servicio de Anatomia Patologica, Hospital Txagorritxu, Vitoria-gasteiz. ESPAÑA
(2) Servicio de Anatomía Patológica del Hospital Txagorritxu ESPAÑA
|
|
Resumen
abortion clinics with sedation wa state abortion clinics with sedation wa state

- Trabajo completo en PDF
|
|
Historia Clínica
Antecedentes maternos: mujer de 35 años, con una gestación anterior normal y parto por cesárea en el año 2004. Recién nacido intervenido por tetralogía de Fallot y onfalocele. No hay otros datos de antecedentes patológicos destacables. Examen físico y analíticas de la madre normales.
 
Feto con OI : nótese el acortamiento de las extremidades y peso y tallos bajos para la edad gestacional |
|
Antecedentes fetales: feto muerto por interrupción voluntaria del embarazo el 07/05/2009. Fecha de la autopsia: 08/05/2009.
Ecografía del II trimestre: feto malformado con acortamiento severo de las extremidades.
Amniocéntesis: cariotipo 46 xx normal.
Examen fisico:
-
Peso: 130 g.
-
Talla: 18,4 cm.
-
PC: 17 cm.
-
PT: 11 cm.
-
Long. pie: 2,7 cm
DX: peso y talla bajos para la edad gestacional.
Acortamiento severo de las extremidades. |
 
Se observan múltiples fracturas costales , que le dan la apariencia a las costillas de estar de estar plegadas.
Examen radiográfico:
-
Cabeza grande con defecto de osificación de la calota.
-
Tórax estrecho
-
Costilla corta con presencia de múltiples callos de fractura.
-
Alteración generalizada de la densidad ósea.
-
Huesos cortos y encorvados.
-
Huesos iliacos redondos.
-
Defecto de osificación del pubis.
-
Platispondilia
DX: Osteogenesis imperfecta tipo AD.
|
|
Examen Microscópico
Histológicamente la OI se caracteriza por una deficiente osificación y mayor cantidad de cartílago. En la unión metafisioepifisiaria los condrocitos tratan de disponerse en columnas en forma regular y ordenada similar al tejido cartilaginoso normal; sin embargo, el tejido que se va depositando en las lengüetas cartilaginosas es un tejido óseo primitivo, escaso e inmaduro. Este tejido es incapaz de formar la trama reticular en que las trabeculas se disponen normalmente y en su lugar se forman islotes discontinuos. Aunque el hueso cortical es hipercelular sus osteoblastos no tienen una actividad normal, formándose por esta razón una cortical delgada y deficiente compuesta por tejido óseo no lamelar (tejido burdo e irregular), con fácil predisposición a las fracturas y remodelación ósea alterada que da lugar al encorvamiento y deformidades que caracterizan esta enfermedad.

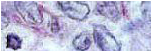
Examen histologico: 1 metacarpiano , feto con OI. Se observa una disposicion irregular de los condrocitos , y el tejido oseo peculiar caracteristico de la OI .

Examen histologico : feto normal. Observar la diferencia en la disposicion de los condrocitos , que se agrupan en forma ordenada formando columnas , y notese el tejido oseo espicular normal en muestra tomada de el mismo sitio en un feto de igual edad gestacional , que el de nuestro caso clinico.

Mano feto con OI. Observese el retraso en la formacion de los centros de osificacion.

Quinto metacarpiano , feto con OI. Observar la delgadez de la cortical y la ausencia enla formacion de trabeculas oseas.

Mano feto 20 semanas normal, observar el tejido oseo presente ya en la mayoria de los centros de osificacion.

Quinto metacarpiano , feto normal. Observar la casi completa osificacion y la conversion de la espicula osea para formar la homogenea disposicion de las trabeculas.

Pie feto con OI.

Primera metatarso falange de feto con OI. No hay formacion de tespiculas ni de tejido oseo normal.
.jpg)
Pie feto normal. Articulacion peroneo -astragalina y hueso calcaneo.

Primera metatarso falange de feto normal.

Primera metatarso falange de feto normal. Formacion de espiculas oseas que dan lugar trabeculas homogeneas .

Calota feto con OI.

Calota feto normal

Clavicula feto con OI

Clavicula feto normal.

Costilla feto con OI.

Costilla feto normal.

Vértebra dorsal feto con OI.

Vértebra dorsal feto con OI.

Vértebra dorsal feto normal.

Vértebra dorsal feto normal. Detallese la disposicion en columnas y ordenada de los condrocitos y la formacion de la espicula osea normal.
|
|
Comentarios
Osteogénesis imperfecta también llamada niños de cristal o enfermedad de los huesos rotos, definida como un grupo heterogéneo de anormalidades hereditarias. Esta enfermedad es producida por mutaciones en los genes COL1A1 (crom17) y COL1A2 (crm7) que codifican las dos cadenas de pro-a1 y una cadena de pro-a2, que entrelazadas en una triple hélice forman el tipo I procolágeno el cual da lugar al colágeno tipo I, que es la principal proteína estructural de la matriz del hueso, los tendones, escleras, cornea, dentina y piel.
La OI es una enfermedad rara. Su incidencia se estima entre 1:10000 y 1:15000 como límites inferiores ya que las formas más severas frecuentemente no sobreviven al nacimiento y las leves algunas veces no se diagnostican por presentar inadvertido cuadro clínico. En el mundo solamente un 0,008% de la población mundial esta afectada por OI (0,5 millones de afectados) y en España se calculan un mínimo de 2700 afectados.
Las características fenotipicas de la OI varían ampliamente desde individuos con muy baja estatura, múltiples fracturas, severas deformidades óseas, dentinogensis imperfecta, escleras azules, pérdida de la audición hasta las formas muy leves asintomáticas con estatura normal y leve predisposición a fracturas sin otros hallazgos. La OI ha sido clasificada en cuatro tipos basados en la presentación clínica, radiología, historia familiar y natural y estudios genéticos moleculares.
|
I Leve
50% |
Normal o acortamiento
leve, fragilidad y deformidad ósea,
escleras azules, dentinogénisis
imperfecta |
AD |
Producida por una
disminución en la
producción de pro-a1
procolageno |
|
II Perinatal
Letal
5% |
Letal, mínima mineralización del
cráneo ,extrema osteoporosis, fragilidad y
deformidad ósea marcada |
AD |
Resultan de mutaciones
en las cadenas pro-a1 y
pro-a2, la mas común
es la sustitución de
un aminoácido
importante de la triple
hélice por un residuo
de glicina, lo que
provoca su inestabilidad
estructural y defectuosa
formación del colágeno
tipo I. |
|
III Progresiva
Deformante
20 % |
Deformidad severa y progresiva de los
huesos, baja estatura, tórax en barril,
múltiples problemas respiratorios, escleras
azules, dentinogenesis imperfecta y
frecuentemente pérdida de la audición. |
AR |
|
IV Moderadamente
Severa
24 % |
En cuanto a la gravedad se clasifica entre
I y III, desde: leve a moderada deformidad
ósea , estatura variable , con frecuencia
dientes frágiles, puede haber perdida de la
audición, y las escleras suelen ser blancas. |
AD |
|
|
Diagnostico prenatal
Se realiza cuando existe sospecha por antecedentes familiares o gestaciones anteriores con OI. Las pruebas que se realizan son:
1. El ultrasonido: en OI tipo II desde la 14 semana y OI tipo III desde la 16 semana examinando el esqueleto del feto, desviaciones, fracturas u otras anormalidades, aunque las formas mas leves no suelen apreciarse hasta el nacimiento.
2. Examen del tejido corionico: suele realizarse de la 10 a 14 semanas examinándose las células de la placenta y en algunas circunstancias se pueden descubrir proteínas anormales de colágeno o una mutación genética que indica OI.
3. Amniocentesis: se realiza entre la 15 a la 18 semana examinando las células fetales de descamación en el líquido amniótico que llevan toda la carga genética que ha heredado el feto, útil para buscar una mutación genética que haya originado OI.
|
|
Diagnostico posterior al nacimiento
Se realiza cuando se sospecha de OI por antecedentes familiares, fracturas mínimas en ausencia de traumas u otros desordenes óseos; baja estatura, escleras azules, dentinogénesis imperfecta, hipoacusia o sordera, entre otros.
El diagnóstico puede hacerse sólo clínicamente o en ocasiones suele necesitarse otros exámenes como: mediciones radiográficas, densitometría, análisis del colágeno sintetizado in Vitro por los fibroblastos dérmicos mediante muestra obtenida por biopsia de piel; evaluación histomorfometrica de muestra de la cresta iliaca. También suele necesitarse pruebas genéticas que detecten y analizen las mutaciones en COL1A1 y COL1A2, y clasifiquen claramente la OI.
|
|
Tratamiento
No existe, hoy en día, tratamiento curativo de la enfermedad. El tratamiento va dirigido a la prevención y control de los síntomas, maximizar la movilidad y desarrollar una masa ósea óptima. Los tratamientos más utilizados son los bifosfonatos y el pamidronato que disminuyen la resorción ósea, el dolor y ayudan al remodelamiento del hueso. Los tratamientos ortopédicos que consisten en la fijación interna con barras intramedulares que mejoran la movilidad y la deformidad ósea.
Otras tratamientos que se encuentran en estudio son la terapia celular que consiste en el trasplante de medula ósea de un donante normal al paciente con OI, y la terapia génica que consiste en el trasplante autólogo de células estromales de medula ósea previamente modificadas genéticamente suprimiéndoseles o silenciándoles el gen mutante por un gen de colágeno normal.
En conclusión la terapia génica efectiva en la OI requerirá la supresión del alelo del colágeno mutante sin afectar el normal alelo endógeno; sin embargo, la alta heterogenicidad de las mutaciones en la OI requiere el desarrollo de estrategias de mutaciones independientes que puedan cumplir un amplio espectro en la OI.
|
|
Bibliografía
Orthopaedic pathology, second edition, Lippincott Williams & Wilkins, Vincent J. Vigorita, Osteogénesis imperfecta, pages 200-206.
Enfermedades metabólicas, degenerativas e inflamatorias de los huesos. H. L. Jafre. Cap. 6, Osteogénesis imperfecta, pag. 173-186.
GeneReviews, Osteogenesis Imperfecta, D Steiner Robert, Oregon Health and Science University, 2005.
Gene Therapy, Gene therapy approaches for osteogenesis imperfecta, C Niyibizi, S Wand, Z Mi and PD Robbins, 2004, 11, pages 408-416.
Firestein: Kelley`s Textbook of Rheumatology, 8th ed. 2008. Deborah Krakow. Pages. 1 - 47.
Dormans: Pediatric Orthopedics and Sports Medicine: Requisites, 1st ed. 2004, Osteogenesis imperfecta.
The New England Journal of Medicine, Targeting Gene Therapy for Osteogenesis Imperfecta, Darwin J. Prockop. M.D., Ph.D. Number 22, 2004.
Nelson Textbook of Pediatrics, Section 3, The Skeletal Dysplasias from Kliegman. Pages. 1-2.
Discapnet. Salud, Osteogenesis imperfecta, http://salud.discapnet.es
Genetics Home Reference, Nov. 2007, http://ghr.nlm.nih.gov
Adam: Grainger & Allinson`s Diagnostic Radiolgy, 5th ed., chapter 49 - Metabolic and Endocrine Skeletal Disease, Judith E. Adams. 2008.
|
|

 COMUNICACIONES
COMUNICACIONES