|
Tumoración córticoadrenal con recuento mitótico elevado en un lactante: a propósito de un caso.
Sonia García Hernández[1], Carmen Nieves Hernández León[1], Rosa Nieves Rodríguez Rodríguez[2], Hugo Álvarez-Argüelles Cabrera[1], María del Carmen Martín Corriente[1], Leticia Melgar Vilaplana[1], Antonio Isaac Martín Herrera[1]
(1) Hospital Universitario de Canarias. La Laguna. Tenerife. ESPAÑA
(2) Hospital Universitario de Canarias.
La Laguna. Tenerife. ESPAÑA
|
|
RESUMEN:
sertraline side effects alcohol sertraline and alcohol side effects jaysmith.us Introducción:
Los tumores adrenocorticales en la población pediátrica son raros. Las manifestaciones clínicas y el comportamiento biológico de estas lesiones pueden ser bastantes distintas de lesiones histológicamente similares en la población adulta, haciendo que los criterios patológicos para distinguir tumores benignos de malignos sean equívocos. El patólogo se encuentra con la dificultad de determinar si se encuentra ante una tumoración corticoadrenal benigna o maligna, con el añadido de que los carcinomas muestran una pobre supervivencia a los 5 años.
Presentación del caso:
Presentamos el caso de un lactante con síndrome de Cushing y virilización que mostraba una masa adrenal izquierda en la ecografía abdominal y ACTH basal baja en la analítica de orina. Se le interviene quirúrgicamente.
-Estudio Macroscópico: Glándula suprarrenal izquierda aumentada de tamaño y peso (5 x 4,5 x 4cm y 40gramos), nodular, de superficie bien delimitada y aparentemente encapsulada. Al corte, muestra consistencia algo blanda y planos de sección blanquecinos.
-Estudio Microscópico: Proliferación tumoral encapsulada, que adopta un patrón de crecimiento fundamentalmente en nidos, dispuestos entre una red extensa de capilares. Las células presentan citoplasma eosinófilo y núcleo ovoide, con nucléolos poco evidentes. El recuento de mitosis es elevado por zonas (13 mitosis/20CGA).
-Diagnóstico: Adenoma de la corteza adrenal.
Discusión:
La mayoría de los tumores en niños se diagnostican siguiendo los criterios de Weiss. Basado en esto, muchos tumores adrenocorticales pediátricos serían diagnosticados como carcinomas, pero tendrían un comportamiento biológico benigno, sin recurrencia tumoral o metástasis en un periodo de seguimiento amplio, por lo que es razonable pensar que fueran benignos. Los tumores pediátricos usualmente tienen índices proliferativos más elevados comparados con los tumores en adultos, no significando esto que sean lesiones malignas. En las neoplasias endocrinas, no hay ningún rasgo histomorfológico que sea diagnóstico de malignidad por sí solo, sobretodo en la población pediátrica.
|
|
INTRODUCCIÓN:
Los tumores adrenocorticales son todavía más raros en la población pediátrica que en la adulta, comprendiendo aproximadamente un 0,2% de los tumores pediátricos. El carcinoma adrenocortical es todavía más raro, siendo su incidencia solo de un 0,3 por millón. Hay una predominancia del sexo femenino (niña: niño, 2 a 1) y con una media de edad de 5 años (oscilando de 6 meses a 19 años). En cuanto a la edad de distribución, se describe un grupo infantil (con un pico de incidencia en el primer año de vida), y un grupo adolescente con un pico que abarca de los 9 a los 16 años. En una revisión de Lack et al, parece que hay un cierto predominio de la glándula derecha frente a la izquierda. Los tumores adrenocorticales bilaterales son extremadamente raros, lo mismo que las localizaciones ectópicas.
Muchos tumores pediátricos que son clínicamente benignos tienen hallazgos morfológicos que, comunmente se asocian con malignidad en tumores de adultos. Dado que los tumores corticoadrenales en niños son raros, es difícil sacar conclusiones definitivas sobre el impacto pronóstico de los hallazgos morfológicos. La experiencia limitada de la mayoría de los patólogos con estos tumores puede contribuir a la tendencia a sobrediagnosticar estos raros tumores como un carcinoma. Según esto, el patólogo se encuentra con la dificultad de determinar si se encuentra ante una tumoración corticoadrenal benigna (adenoma) o maligna (carcinoma), con el añadido de que los carcinomas muestran una pobre supervivencia a los 5 años.
|
|
PRESENTACIÓN DEL CASO:
Presentamos el caso de un lactante, varón, de 6 meses de edad, cuya familia consulta a su pediatra de zona por obesidad generalizada llamativa sin relacionarse con aumento de las ingestas, aumento progresivo del vello en la espalda y cara, junto a la presencia de vello suprapúbico. Ante la sospecha de Síndrome de Cushing y virilización se le realiza una ecografía abdominal que pone de manifiesto una masa renal izquierda de 5 x 4 cm, bien delimitada, sin calcificaciones en su interior, ni lesiones hepáticas acompañantes. La analítica de orina muestra ACTH basal baja, cortisol elevado y delta-4 androstendiona elevada. La tensión arterial es normal. En el TC corporal total no se pone de manifiesto invasión vascular adyacente a la masa. La gammagrafía ósea con Tc 99 no detecta captaciones patológicas que indiquen metástasis óseas. Se opera mediante laparoscopia.
-Estudio macroscópico: Se obtiene una glándula suprarrenal izquierda aumentada de tamaño y peso (5 x 4,5 x 4 cm y 40 gramos), nodular, de superficie bien delimitada y, aparentemente encapsulada. Al corte, muestra consistencia algo blanda y planos de sección blanquecinos. Macroscópicamente, no se identifican áreas de hemorragia ni necrosis (Fig 1).
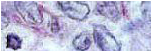
-Estudio microscópico: Se pone de manifiesto una proliferación tumoral encapsulada, que adopta un patrón de crecimiento fundamentalmente en nidos, dispuestos entre una red extensa de capilares (Fig 2). Las células presentan citoplasma eosinófilo y núcleo ovoide, con nucléolos poco evidentes (Fig 3). El recuento de mitosis fue de 13 en 20 CGA, no identificándose mitosis atípicas (Fig 4). No se identifican bandas anchas fibrosas, áreas de necrosis, hemorragia o calcificación distrófica en los cortes estudiados. Tampoco se puso de manifiesto invasión vascular o capsular. Las células tumorales se tiñen con pan-citoqueratina (pan-CK), siendo negativas para la vimentina. El índice proliferativo (Ki-67) es elevado por zonas (Fig 5).
-Diagnóstico: Con todos estos hechos, establecemos el diagnóstico de Adenoma de la corteza adrenal.

Fig 1 -

Fig 2 - HE

Fig 3 - HE

Fig 4 - HE

Fig 5 - Ki 67
|
|
DISCUSIÓN:
Al contrario que la población adulta, la tasa de tumores funcionantes en la población pediátrica es de un 80%. Menos de un 10% son asintomáticos al diagnóstico. La mayoría de los niños presentan virilización, seguido por Síndrome de Cushing, que a menudo se presenta junto a la virilización (60%); un Síndrome de Cushing puro es infrecuente. La obesidad en los niños tiende a ser generalizada en vez de troncular. La feminización es inusual, así como el hiperaldosteronismo primario. Los tumores adrenocorticales que segregan andrógenos exclusivamente son infrecuentes. Los rasgos de la virilización en las niñas incluyen aumento de la masa muscular (hábito hercúleo), así como clitoromegalia, presencia de pelo facial, voz grave, y la aparición de vello púbico. El exceso de andrógenos en niños causa precocidad sexual con alargamiento del pene y presencia de vello púbico (Tabla 1).
Macroscópicamente, los tumores adrenocorticales varían ampliamente en tamaño (de 2,4 a 19 cm) y peso (de 18 a 6000 gramos). Puede haber áreas de hemorragia y necrosis.
La diferenciación histológica entre tumores adrenocorticales benignos y malignos se basa actualmente en los criterios de Weiss et al. (Tabla 2), en la tinción inmunohistoquímica para vimentina y en el índice proliferativo, pero el valor pronóstico de estos criterios en los casos pediátricos son todavía poco claros. La necrosis tumoral, la invasión vascular y capsular, tres apartados de los criterios de Weiss, así como el gran tamaño de la tumoración se relacionan con malignidad y peor pronóstico. Por otra parte, el recuento de mitosis y las mitosis atípicas, dos criterios mayores para Weiss, así como la tasa de proliferación (indicado por el Ki-67 y la tinción positiva para p53) y la inmunotinción para vimentina, parecen irrelevantes para el comportamiento clínico futuro de los tumores en la población pediátrica. Vargas et al. concluyeron que la tinción para p53 y el índice proliferativo (Ki-67) podrían ser de interés para diagnosticar cacinomas adrenales, así como para predecir su comportamiento clínico, pero los tumores pediátricos usualmente tienen índices proliferativos más elevados comparados con los tumores en adultos. Cagle et al. reportaron que los tumores pediátricos tienen más probablemente mitosis, necrosis y pleomorfismo nuclear comparados con los adultos.
|
|
CONCLUSIONES:
En los adultos, la diferenciación entre adenoma y carcinoma está bien establecida según los criterios de Weiss, así como la inmunotinción para vimentina y tamaño tumoral. Sin embargo, no se han establecido criterios diagnósticos claros para los tumores adrenocorticales pediátricos. Es más, la mayoría de los tumores en niños se diagnostican siguiendo los criterios de Weiss. Según esto, muchos tumores adrenocorticales pediátricos serían diagnosticados como carcinomas, pero tendrían un comportamiento biológico benigno, sin recurrencia tumoral o metástasis en un periodo de seguimiento amplio, por lo que es razonable pensar que, en realidad fueran benignos. Para algunos autores, el uso de los criterios de Weiss para adultos puede ser inapropiado para el diagnóstico diferencial de los tumores adrenocorticales en niños.
Es importante señalar que no hay ningún rasgo histológico que por si solo sea diagnóstico de malignidad en los tumores de la corteza adrenal. Especialmente en la población pediátrica, en el diagnóstico de neoplasias endocrinas se deben considerar toda una serie de rasgos histomorfológicos (Tabla 3) antes de que se haga el diagnóstico de carcinoma de corteza adrenal. Los únicos hechos que se pueden tomar como certeros de malignidad adrenal primaria son la presencia de enfermedad recurrente o metastásica.
Wieneke et al. describieron una serie de rasgos o criterios macroscópicos y microscópicos de malignidad para los tumores corticoadrenales en la población pediátrica que cuando se agregan, parece que predicen un comportamiento biológico más agresivo. Cuantos más rasgos se vean, mayor tendencia a la malignidad de la lesión (Tabla 3).
|
|
TABLAS:
Tabla 1:
CLASIFICACIÓN FUNCIONAL DE LOS TUMORES ADRENALES CORTICALES:
1. Hiperfunción Endocrina (hipercorticalismo):
-Hipercortisolismo (Sdme. Cushing)
-Hiperaldosteronismo (Sdme. Conn)
-Virilización
-Feminización.
-Síndrome endocrino mixto
2. No funcional o no hiperfuncional
3. Estatus funcional desconocido
Tabla 2:
CRITERIOS DE WEISS para distinguir neoplasias adrenocorticales benignas de malignas:
1. Alto grado nuclear. Criterios de Fuhrman
2. Más de 5 mitosis en 50 CGA
3. Mitosis atípicas
4. < 25% de las células tumorales son células claras
5. Arquitectura difusa (≥ 33% de las células tumorales)
6. Necrosis
7. Invasión venosa (músculo liso en la pared)
8. Invasión sinusoidal (no músculo liso en la pared)
9. Invasión capsular
La presencia de 3 o más criterios se correlaciona altamente con un comportamiento maligno ulterior.
Tabla 3:
CRITERIOS HISTOLÓGICOS DE MALIGNIDAD PROPUESTOS PARA TUMORES ADRENOCORTICALES EN LA POBLACIÓN PEDIÁTRICA:
-Peso del tumor mayor de 400 g
-Tamaño del tumor mayor de 10,5 cm
-Extensión en tejidos blandos periadrenales y/o órganos adyacentes
-Invasión de la vena cava
-Invasión venosa
-Invasión capsular
-Presencia de necrosis tumoral
-Más de 15 mitosis por 20 CGA (400x)
-Presencia de mitosis atípicas
|
|
Bibliografía
- Cagle PT, Hough AJ, Pysher TJ, et al. Comparison of adrenal cortical tumors in children and adults. Cancer 1986; 57: 2235-7.
- Ernest E. Lack, MD. Tumors of the Adrenal Glands and Extraadenal Paraganglia. AFIP Atlas of Tumor Pathology. Fourth Series. Fascicle 8. 2007.
- Jacqueline A. Wieneke, Lester D.R. Thompson and Clara S. Heffess. Adrenal Cortical Neoplasms in the Pediatric Population. A Clinicopathologic and Immunophenotypic Analysis of 83 patients. The American Journal of Surgical Pathology. 27(7): 867-81, 2003.
- Lucon AM et al. Adrenocortical tumors: results of treatment an study of Weiss´s score as a prognostic factor. Rev Hosp. Clín. Fac. Med. S. Paulo 57 (6): 251-256, 2002.
- Ronald A DeLellis, Ricardo V. Lloyd, Philipp U. Heitz, Charis Eng. WHO Classification. Tumours of Endocrine Organs. IARC. 2004.
- Yoko Misu, Shi-Xu Jiang, Yukifumi Yokota, Masahiko Shibata, Osamu Shinohara, Toru Kameya and Nobuo Matsuura. Clinicopathological Features of Pediatric Functional Adrenocortical Carcinoma Diagnosed by Weiss Criteria; An Analysis of Four Cases. Clin Pediatr Endocrinol 2001; 10(2), 141-146.
|
|

 COMUNICACIONES
COMUNICACIONES