
ISBN: 978-84-692-76778
 COMUNICACIONES
COMUNICACIONES
Nº 1980. Patología Quirúrgica
|
Paraganglioma retroperitoneal: a propósito de un caso. CRISTINA MURILLO LÁZARO[1], LUCÍA M. GONZALEZ LÓPEZ[1], MARCIAL GARCÍA ROJO[1], MARGARITA DELGADO PORTELA[1], JESÚS GONZALEZ GARCÍA[1], FRANCISCO MARTIN DÁVILA[1], RAFAEL LÓPEZ PÉREZ[1], MANUEL CARBAJO VICENTE[1] |
|
buy abortion pill barcelonabuy abortion pill barcelona linkpolyethylenepolyethylene saniblog.chprogesterone ovuleprogesterone tauxEl paraganglioma retroperitoneal no funcionante (no secretor de catecolaminas) es un tumor infrecuente derivado de células cromafines de la cresta neural que asienta en la cadena simpática siendo difícil su diagnóstico pre-operatorio. A continuación se describe el caso de un varón de 42 años que ingresa por sintomatología gastrointestinal y descompensación hepática por VHC, al que se le descubre tras la realización de una ecografía, una masa sólida en el retroperitoneo izquierdo. Se efectúa biopsia preoperatoria produciéndose tras su realización un gran hematoma. Se procede a la extirpación quirúrgica de urgencia, resecando una masa de 5 cm de diametro máximo, bien delimitada y encapsulada, mostrando una superficie de corte hemorrágica de coloración marrón-rojiza. En los cortes histológicos se observa una proliferación neoplásica compuesta por nidos y trabéculas de células con escasa atipia y con marcada trama vascular positivas para cromogranina, sinaptofisina y neurolasa específica. Las células sustentaculares fueron positivas para la S-100. Los criterios de malignidad del paraganglioma son controvertidos considerando algunos la metástasis el único criterio válido. El tratamiento de elección es la cirugía.
|
||
|
|
El paraganglioma es un tumor derivado de las células cromafines de la cresta neural. Se divide en tumores que derivan del sistema parasimpático o del sistema simpático (1). Los tumores que derivan del sistema nervioso parasimpático se localizan exclusivamente en base de cráneo y cuello y asientan entre el cuerpo carotídeo y globus yugulotimpánico, siendo típicamente no secretor de catecolaminas. Por el contrario muchos de los tumores derivados de los ganglios simpáticos se localizan en el abdomen y la mayoría suele producir un exceso de catecolaminas incluyendo síntomas como palpitaciones, dolor de cabeza, hipertensión arterial.. Si se produce en la médula suprarrenal se denomina feocromocitoma. En el resto de paraganglios simpáticos se denomina paraganglioma extra-adrenal, localizándose el 5% en base de craneo y cuello, 10% en vejiga y próstata. Los que se localizan a lo largo de la aorta, un 10% asienta en tórax y un 75% en abdomen localizándose la mayoría en el órgano de de Zuckerkandl(1,2,3) La mayoría de paragangliomas se diagnostican entre los 30 y 45 años, sin embargo las formas malignas se diagnostican a una edad media más temprana (4,5) Los hombres y mujeres están afectados por igual. La presentación del paraganglioma es variada: produciendo efecto masa o dolor abdominal, como un hallazgo incidental, o por producción de exceso de catecolaminas. Su identificación y adecuado tratamiento puede evitar la morbilidad y potencial mortalidad asociada sobre todo si es sintomático. El paraganglioma no funcional, como parece ser nuestro caso, rara vez se diagnostica antes del pre-operatorio.(6)
|
|
|
|
Varón de 42 años, adicto a drogas, que ingresa por un cuadro de gastroenteritis aguda y descompensación de su cirrosis por VHC (ascitis, hipertensión portal ) Tras la realización de una ecografía abdominal se descubre una masa sólida de 5x4 cm, en el espacio paraórtico izquierdo, sin relación con la glándula suprarrenal, que comprime los segmentos gastrointestinales (produciendo síntomas gastrointestinales por compresión, efecto masa).Se sospecha que pudiera tratarse con estos hallazgos en primer lugar de un GIST. Se realiza una biopsia preoperatoria produciéndose clínica de abdomen agudo con caída del hematocrito, debido a la formación de un gran hematoma de 18x14x13 cm que desplaza grandes vasos. Se realiza extirpación de la tumoración de carácter de urgencia, observando tras la resección que no tiene relación con asas intestinales.
|
|
|
|
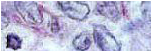
Macroscópicamente se trata de una formación nodular grisácea encapsulada de 5 cm de diámetro máximo y 55,7 gr de peso. Al corte la superficie es de coloración marronácea con áreas hemorrágicas y algún área fibrótica (figura 1). En los cortes histológicos se observa una proliferación neoplásica constituida por nidos y cordones de células pequeñas poligonales o fusocelulares con escasa atipia, de citoplasma eosinófilo y núcleo hipercromático (figuras 2,3). El índice mitótico (ki-67) es del 10%. Mediante técnicas de inmunohistoquímica se observa positividad citoplásmica para cromogranina (figura 4), sinaptofisina y enolasa neuronal específica (figura 5) en las células neoplásicas y positividad par la S-100 en las células sustentaculares (figura 6).
|
|
|
|
La nomenclatura del paraganglioma ha sido controvertida. En la clasificación de la WHO del 2004 de los tumores endocrinos, se define como feocromocitoma un tumor derivado de las células cromafines de la médula adrenal. Los tumores derivados de los paraganglios del sistema simpático y parasimpático extra-adrenal se denominan paragangliomas extra-adrenales (7). El paraganglioma retroperitoneal deriva del sistema nervioso simpático y la mayoría se localiza en el órgano de Zuckerkand (a la izquierda de la aorta cerca de la arteria mesentérica inferior) o entre la aorta y la vena renal (9,11). El paraganglioma y el feocromocitoma se asocian a diversos desordenes genéticos como el MEN2A Y MEN2B, la enfermedad de von Hippel-Lindau, la neurofibromatosis tipo 1 y la triada de Carney (11). Recientemente se ha descrito un síndrome familiar de paraganglioma resultado de la mutación del gen succinato deshidrogenasa (12). Estos tumores pueden secretar epinefrina y norepinefrina que se traducen clínicamente con hipertensión, dolor de cabeza, palpitaciones y sudoración. El paranglioma retroperitoneal secreta catecolaminas (usualmente norepinefrina) entre un 15-60% de casos. En nuestro caso debido a la ausencia de síntomas clínicos no se realizó cuantificación de catecolaminas ya que no se sospechaba un paraganglioma inicialmente. El diagnóstico ante la sospecha de paraganglioma se realiza con la medición de catecolaminas ,bien cuantificando la concentración de estas en orina de 24 horas o los niveles de metanefrina libre en plasma, que según algunos estudios por su sensibilidad y especificidad es el test de elección.,(9). Se debe de acompañar de técnicas de imagen (TAC abdominal, resonancia magnética ) para su localización siendo difícil su diagnóstico en ausencia de síntomas. Los paragangliomas retroperitoneales suelen ser tumores nodulares, parcial o completamente encapsulados y de consistencia firme. La superficie de corte suele ser de coloración rojo-marronácea, vascularizada, homogénea y ocasionalmente fibrótica. En los cortes histológicos puede parecerse a los típicos paragangliomas de cabeza y cuello con discretos pequeños de nidos de células (patrón alveolar o Zellballen), sin embargo es más característico un patrón de trabéculas anchas anastomosadas con marcada trama vascular, disponiéndose las células adyacentes al endotelio vascular. En ocasiones el patrón que se observa es sólido. Las células muestran un citoplasma eosinófilo y núcleo hipercromático. Se pueden identificar glóbulos eosinófilos en el citoplasma de las células tumorales (considerado criterio de benignidad por Linnoila como se comenta a continuación). Se puede evidenciar hemorragia y congestión. Ocasionalmente pueden mostrar gran pleomorfismo ya que se compone de células fusocelulares con escaso citoplasma y gran núcleo e incluso pierden el patrón de organización típico. La mitosis suelen ser escasas. Se ha descrito algún caso que contiene pigmento que sugiere que sea neuromelanina (12). Mediante técnicas de inmunohistoquímico se evidencia en las células tumorales positividad para enolasa neuronal específica, cromogranina, sinaptofisina y proteína neurofilamentosa, rodeadas estas células tumorales por células sustentaculares S-100 positivas (13). La escasez o ausencia de estas células sustentaculares se ha postulado como posible factor de malignidad (14). También se han identificado diversa variedad de neuropéptidos. Mediante microscopia electrónica se puede distinguir los típicos pequeños gránulos densos que contienen catecolaminas y otros componentes neuroendocrinos. El diagnóstico diferencial a tener en cuenta ante una masa retroperitoneal izquierda debe incluir paraganglioma, liposarcoma bien diferenciado, leiomiosarcoma, linfoma, melanoma , si tiene relación con el segmento intestinal un GIST y metástasis. El pronóstico del paraganglioma es muy difícil de definir siendo la metástasis el único criterio de malignidad para algunos (15). Clarke et al postula que una localización extra-adrenal, el sexo masculino, ser joven, el peso del tumor y el ki-67 son factores pronósticos de malignidad (4). Linnoila et al concluyen que una localización extra-adrenal, pérdida de nodularidad , necrosis confluente y ausencia de glóbulos hialinos se asocia a malignidad (presentando el 71% de los paragangliomas malignos dos o tres criterios de estos, mientras que el 89% de los benignos sólo presentan un criterio o ninguno)(16). Parece que la localización del paraganglioma influye en su comportamiento biológico teniendo mayor incidencia de transformación maligna los parangliomas retroperitoneales que por ejemplo un paraganglioma del cuerpo carotideo (7,15) No se ha evidenciado que el grado de actividad catecolaminérgica influya en el grado de malignidad. El paraganglioma maligno metastatiza principalmente a ganglios linfáticos, pulmón, hueso, hígado y ocasionalmente a otros lugares (17). El paraganglioma múltiple debe distinguirse de las metátasis. El paraganglioma retroperitoneal tiene mayor tendencia a metastatizar que otros paragangliomas (18). El tratamiento de elección es la cirugía( 19,20) realizada clásicamente por laparotomía transabdominal . El paraganglioma retroperitoneal puede resecarse también actualmente por cirugía laparascópica bien transabdominal o retroperitoneal. (20, 21). Ante la sospecha no debe realizarse biopsia preoperatoria por el riesgo de crisis hipertensiva si es funcionante ( por lo que si se realiza cualquier manipulación del mismo debe darse en el preoperatorio alfa seguido de beta bloqueadores adrenérgicos)o de sangrado por su alta vascularización. Algunos autores postulan la realización de una embolizacion preoperatoria (discutida por algunos) antes de la resección (11).
|
|
|
|
7 Yau KK, Siu WT, Li MK. Unual Cause of Acute Abdominal- Ruptured Retroperitoneal Paraganglioma. Asian J Surg. 2008 Jan;31(1):32-5. 8 DeLellis RA, Heitz PU, Eng C, eds. Pathology and Genetics of Tumours of Endocrine Organs. Lyon, France: IARC Press; 2004 20 Brewster JB, Sundaram CP. Laparoscopic Resection of an Interaortocaval Paraganglioma: Diagnosis Following a Needle Biopsy. JSLS. 2007 Oct-Dec;11(4):502-5.
|
|
|
|
- Daniel Sanchez Guerra - ESPAÑA (10/11/2009 21:35:39)
- ALEXANDRA GENÉ HEYM - ESPAÑA (13/11/2009 8:37:20)
- María Isabel Oviedo Ramírez - ESPAÑA (15/11/2009 14:18:31)
- Franklin Idrovo Mora - ESPAÑA (15/11/2009 22:14:57)
- MARIO CASTILLO GUTIÈRREZ - MEJICO (16/11/2009 18:00:08)
- CRISTINA MURILLO LÁZARO - ESPAÑA (Autor) (18/11/2009 23:06:45)
- Johamel Ramos Valdés - CUBA (18/11/2009 23:54:27)
- CRISTINA MURILLO LÁZARO - ESPAÑA (Autor) (30/11/2009 23:27:59)
|
|
|
|
|







Web mantenido y actualizado por el Servicio de informática uclm. Modificado: 16/06/2015 11:37:37