
ISBN: 978-84-692-76778
 COMUNICACIONES
COMUNICACIONES
Nº 1867. Patología Quirúrgica
|
Alcaptonuria. Presentación de un caso. Daniel Artiles Martínez[1], Kenia González Valcarcel[1], Nancy Cárdenas Caballero[1] |
|
citalopram alcohol cravingscitalopram and alcohol nhs readamlodipin actavisamlodipin teva bivirkninger
|
||
|
|
El metabolismo de los aminoácidos tirosina y fenilalanina representa una importante encrucijada metabólica de cuya alteración se originan diversas enfermedades (anexo. 1). De las más importantes por su significado clínico y frecuencia, se hallan la fenilcetonuria, la tirosinemia de tipo I y la alcaptonuria (Aciduria Homogentísica). Esta última aunque ya conocida desde el siglo XVI, sirvió para que Archibald Garrod en 1909 empleara por primera vez el término de error congénito del metabolismo y es la primera enfermedad del metabolismo a la que se le atribuye un defecto hereditario autosómico recesivo. (1,2) El ácido homogenístico, es el producto de la degradación del aminoácido tirosina, y este a su vez es transformado por la enzima 1,2-dioxigenasa homogenistica (HGO), a ácido maleil-acetoacético. La interrupción de esta vía conduce a una acumulación en los tejidos del ácido homogenístico, el cual le confiere al tejido conjuntivo una coloración negruzca, de ahí la denominación de ocronosis y de alcaptonuria por su eliminación a través de la orina adquiriendo coloración oscura al oxidarse en el ambiente exterior o al alcalinizarla. (3, 4, 5) La prevalencia es de 1 por cada 100.000-200.000 individuos y en el estudio molecular del gen HGO se detecta la presencia de mutaciones en casi el 100% de los casos (aunque no es imprescindible para el diagnóstico ya que la presencia del ácido homogenístico en la orina es patognomónica de la enfermedad). Este gen se encuentra en el brazo largo del cromosoma 3 (3q21-23) y muestra una gran variedad alélica (hasta le fecha más de 80 mutaciones diferentes). Como se trata de una enfermedad hereditaria, el consejo genético es importante para su prevención. Debe ser diferenciada de la ocronosis secundaria al uso de minociclinas y otras tetraciclinas (3, 6, 7) Desde el punto de vista clínico permanece inadvertida durante años manifestándose solo por orinas de color oscuro, motivo de preocupación y consulta por parte de la madre y si el médico no está informado sobre su significado, pasa inadvertida hasta la tercera o cuarta década de la vida en que la acumulación del pigmento provoca degeneración severa del cartílago. Compromete sobre todo a la columna vertebral, con calcificación de los discos y fusión vertebral. Afecta a grandes articulaciones y respeta típicamente las sacroiliacas. El diagnóstico se basa en la triada de artritis degenerativa, depósito de pigmento en los tejidos mesenquimales y orinas oscuras. (5, 7) El grado de afectación de los pacientes es variable. A nivel de los tegumentos aparecen manchas oscuras en las escleróticas (signo de Osler) y la piel, también en los cartílagos que se trasluce en el cartílago auricular. En pacientes varones es causa de prostatitis y de forma menos frecuente la afectación de válvulas cardiacas sobre todo aórtica. (3, 4).
|
|
|
|
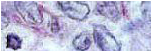
Paciente femenina MVM de 52 años de edad y piel blanca, con antecedentes de haber sido llevada durante su infancia varias veces a consulta por presentar orinas oscuras, lo cual se acrecentaba al dejarlas reposar por varias horas. En esa ocasión se le señaló que estaba en relación con los alimentos. Siguió su evolución natural y cuatro décadas después es valorada por ortopedia por presentar dolores de las grandes articulaciones (hombro, rodillas y columna vertebral) realizándose TAC de columna lumbo-sacra donde se evidenció hernia discal a nivel del espacio L2-L3. Años después asiste a consulta de dermatología por presentar lesión pigmentada de color negro-azulosa de 4 años de evolución localizada en los extremos laterales y mediales de ambas manos (fig-1, 2). Se le practicó biopsia (fig.3) de la lesión en la que se constató a nivel de la dermis superficial y profunda degeneración del colágeno con depósito de un material denso de color ocre el cual fue identificado como pigmento ocronótico, y tras descartarse las causas externas de pigmentación, relacionadas con la actividad doméstica, laboral, cosmética o medicamentosa, se citó a la paciente la cual en el interrogatorio narró la evolución natural de su enfermedad descrita hasta este momento. Además al examen físico se constató coloración oscura del hélix (fig-4) y la esclera (fig-5), cuyos rasgos se acentuaron con la edad. Se solicitó a la paciente que recogiera la micción matutina (500 ml), a la cual se le adicionó ½ ámpula de bicarbonato y se dejó reposar por espacio de 2 horas adquiriendo un manifiesto color negro. Por todo lo anterior se realizó el diagnóstico de alcaptonuria. Fue remitida al servicio de reumatología para que se iniciara el debido tratamiento.
|
|
|
|
La alcaptonuria a diferencia de otras enfermedades metabólicas como la fenilcetonuria o el albinismo, tiene un comienzo insidioso y poco llamativo, siendo la coloración negruzca de la orina, el único síntoma durante décadas y este el motivo de consulta más frecuente en la infancia de estos pacientes. Sin embargo la evolución natural sin intervención terapéutica, puede acarrear con el paso de los años una enfermedad invalidante con significativa afectación de articulaciones importantes y más grave (pero menos frecuente) de la válvula aórtica. Aunque el tratamiento hasta el momento utilizado (abundante líquido para aumentar la excreción del ácido homogenístico y ácido ascórbico para la regeneración del tejido mesenquimatoso dañado) no resuelve la causa básica que es el déficit de la enzima 1,2-dioxigenasa homogenistica (HGO), al menos reduce el avance de la enfermedad y por ende sus secuelas. De ahí la importancia del diagnóstico precoz de la enfermedad, la que por su baja prevalencia y la transmisión autosómica recesiva resulta casi imposible preverla mediante seguimiento de los portadores sanos, al presentarse a saltos en distintas generaciones. En el caso que nos atañe, el diagnóstico se realizó mediante una biopsia de piel, lo que constituye una forma inusual de llegar al diagnóstico según lo que pudimos consultar en la bibliografía revisada. Por lo que no queremos pasar por alto la importancia de reconocer este pigmento natural distinto de la hemosiderina y de la melanina (más por su forma que por sus propiedades tintoriales) ya sea en una biopsia de piel, cartílago o más desafortunadamente en la válvula aórtica en la morgue.
|
|
|
|
1. M Izquierdo, A Avellaneda. Alcaptonuria. www.geosalud.com. Feb. 2004. 2. F Novoa, M Colombo. Errores innatos del metabolismo. Rev Chil Neuro-Psiquiat 2001; 39(1): 25-27. 3. L Ffolkes, D Brull, S Krywawych, M Hayward, S E Hughes. Aortic stenosis in cardiovascular ochronosis. Journal of Clinical Pathology 2007;60:92-93. 4. E Erek, A Casselman, H Vanermen. Cardiac Ochronosis. Valvular Heart Disease with Dark Green Discoloration of the Leaflets. Tex Heart Inst J. 2004; 31(4): 445447. 5. P Suwannarat, Ch Phornphutkul, I Bernardini, M Turner, W Gahl. Minocycline-Induced Hyperpigmentation Masquerading as Alkaptonuria in Individuals With Joint Pain. ARTHRITIS & RHEUMATISM. Vol. 50, No. 11, November 2004, pp 36983701. 6. M Roser, J Moller, T Komoda2, Ch Knosalla2, P Stawowy. Alkaptonuric aortic stenosis. doi:10.1093/eurheartj/ehm406 Online publish ahead of print 7 September 2007. 7. M Ferreira, M Sanches, I Lobo, M Selores. Alkaptonuric ochronosis. EJD, vol. 17, n° 4, July-August 2007.
|
|
|
|
- María Isabel Oviedo Ramírez - ESPAÑA (01/11/2009 16:37:55)
- Teresa Rinaldi Catalá - ESPAÑA (04/11/2009 19:31:15)
- Isidro Machado - ESPAÑA (05/11/2009 11:53:12)
- Carlos Manuel Neira de Paz - ESPAÑA (08/11/2009 21:29:39)
- junkal gallo garmendia - ESPAÑA (12/11/2009 10:24:54)
- Francisco Parrilla Arias - CUBA (20/11/2009 18:11:04)
|
|
|
|
|








Web mantenido y actualizado por el Servicio de informática uclm. Modificado: 16/06/2015 11:37:37